
Ricardo Bustamante B. 1
Rev Chil Anest Vol. 46 Núm. 3 pp. 116-130|https://10.25237/revchilanestv46n03.03
PDF|ePub|RIS
Los anestesiólogos hemos administramos anestesia durante casi 100 años sin el uso de los mal llamados relajantes musculares (desde Morton en 1846, hasta Griffith en 1942). De modo que pudimos vivir un siglo sin estas drogas tan relacionadas con nuestra especialidad y probablemente las hemos subutilizado en algunas oportunidades y las hemos usado demás en otras. Digo mal llamados relajantes musculares, debido a que la denominación es bastante universal, pues el nombre más apropiado es el de bloqueadores neuromusculares (BNM), para diferenciarlos de aquellos fármacos que actúan a nivel del sistema nervioso central. Si fuéramos más puristas, este nombre tampoco es del todo adecuado, porque la succinilcolina, uno de sus más conspicuos representantes, no bloquea receptores y por lo tanto, no es un bloqueador, aunque sin lugar a dudas produce relajación de la musculatura estriada.
La introducción de los BNM a la práctica clínica cambió la forma de definir la anestesia. Antes de su existencia, la intubación endotraqueal era una excepción y si se necesitaba relajación muscular, se obtenía aumentando la fracción inspiratoria de anestésicos inhalatorios, mucho menos seguros que los actuales y con evidente riesgo de depresión respiratoria y cardiovascular. Después que el uso de BNM se hizo habitual, se produjo un cambio conceptual y la anestesia se definió como una tríada formada por tres elementos inseparables: narcosis, analgesia y relajación muscular 1 .
La historia de los bloqueadores neuromusculares puede dividirse en dos períodos: la historia antigua, referida fundamentalmente al descubrimiento e investigaciones relacionadas con el curare y la historia moderna, referida al desarrollo de moléculas que fueron cada vez más específicas en su acción y con un cada vez mejor margen de seguridad terapéutico.
La Fascinante Historia del Curare
La historia del curare abarca unos 300 años, desde la llegada de los aventureros que siguieron a Colón después de su viaje al Nuevo Mundo, las descripciones de la muerte tras recibir una flecha o un dardo disparado por los nativos hecha por Sir Walter Raleigh y, su traslado a Europa por los exploradores para investigar lo que producía la muerte 2 . Se trata de una historia romántica de exploración, curiosidad personal, conjeturas visionarias y cierto grado de serendipia 3 .
El contacto de Colón con aborígenes que usaran curare se ha prestado para muchas especulaciones, pero ninguna evidencia concreta 4 . Después de la llegada de Colón al Nuevo Mundo se establecieron colonias en la isla La Española (actualmente Haití y República Dominicana) y Darién, al sur del istmo de Panamá. De allí surgieron expediciones al norte (México) y al interior del continente sudamericano. El descubrimiento de oro, especialmente por las exploraciones de Pizarro, llegó a Europa como la noticia del oro de California unos siglos después, por lo que muchos navegantes sin gran preparación intentaron atravesar el entonces llamado mar Océano, algunos con éxito, mientras que otros, desconocedores de los problemas de los largos viajes y de los vientos del sur, nunca llegaron a su destino.
Los que llegaron, se vieron sometidos al ataque de nativos hostiles que emergían de una jungla impenetrable, armados con lanzas y dardos embebidos en cierta substancia de propiedades místicas y obviamente mortales (Figura 1). Este sistema no era nuevo; desde tiempos bíblicos, cazadores y guerreros untaban sus flechas con sustancias envenenadas para hacerlas más mortales. Había sido descrito por Virgilio en la Eneida y luego, en la Edad Media, utilizado por celtas y galos, pero reemplazado por otras armas más eficientes, como la pólvora, en Europa y Asia.

Figura 1. Las flechas, lanzas y dardos de algunas tribus de Latinoamérica eran impregnadas con un veneno que los españoles llamaron curare, producto de una mezcla de varias sustancias vegetales (entre ellas el Chondrodendron tomentosum) y animales.
Las descripciones de las muertes ocasionadas por las flechas envenenadas en Sudamérica parecieron en Europa, sobre todo contadas por ansiosos conferencistas en busca de éxito, un acto de brujería. Por supuesto, muchas de estas historias eran adornadas para impresionar a una audiencia ansiosa, incapaz de discernir entre lo real y lo fantástico. Estas muertes fueron especialmente frecuentes entre los exploradores que se acercaban al río Amazonas y a la cuenca del Orinoco, donde los sanguinarios guerreros nativos de estas regiones utilizaban un veneno particularmente potente para exterminar a sus presas de caza y a sus enemigos.
Los conquistadores observaron que los nativos embebían sus dardos y flechas en un brebaje pegajoso, probablemente compuesto por hierbas, cuyo efecto letal era obvio. La muerte que causaba era realmente horrenda. Uno de los más espantosos aspectos de la naturaleza de este tipo de muerte era que parecía no haber nada para impedirla. Era lógico que pudiera existir un antídoto y, al parecer los nativos lo conocían, pero antes había que conocer qué era el veneno y como mataba a las víctimas. Este veneno era obtenido a partir de ciertas hierbas y raíces de un bejuco (enredadera) que prontamente fue llamado “hierba” por los españoles.
Los primeros detalles fueron descritos por el monje italiano Martín de Angleira, que relatara en la corte española hacia 1514, (sin haber estado nunca en América), que la pócima venenosa era hecha por una pequeña secta de ancianas que la mantenían sellada por un par de días y que a menudo morían al inhalar sus gases5. Su composición y manufactura era mantenida en secreto. Un informe posterior de Nicolás Bautista Monardes, que tampoco aparece en las listas de los viajeros a América, pero que fuera un destacado médico sevillano, autor de varias obras de gran valor y por algunos considerado como el padre de la farmacología, también reporta sobre el peligro de la inhalación del preparado y de la horrible muerte que causaba, describiendo todo un ritual que envolvía su preparación, rodeando el relato de magia y brujería. El trabajo específico se llama “Dos libros, el uno que trata de todas las cosas que se traen de nuestras Indias Occidentales, que sirven al uso de la medicina, y el otro que trata de la piedra bezaar, y de la yerva escuerçonera”, publicado en Sevilla en 1569 por Hernando Díaz.
Sin embargo, las descripciones más dramáticas del efecto del “ourari”, como lo llamó uno de sus tenientes, fueron hechas en 1595 tras el regreso a Europa de Sir Walter Raleigh desde Guinea (Guyana), en que algunos miembros de su expedición penetraron por la cuenca del Orinoco, incluido su hijo que perdió la vida. Las personas que recibían las flechas envenenadas eran víctimas de un tormento insoportable: permanecían conscientes, con la vista fija, pero eran incapaces de gritar o llorar durante su agonía. Los ingleses también creían que existía un antídoto, pero que ni siquiera los españoles empleando torturas, a las cuales estaban acostumbrados a usar con los nativos (y con sus congéneres europeos que pensaban distinto), lograron obtener.
Las descripciones del corsario inglés también se prestan a dudas pues, aunque educado en Oxford, sus vívidas descripciones de El Dorado, ponen un manto de inseguridad sobre sus reales intenciones o su veracidad. Raleigh, probablemente informado por sus hombres y por los españoles, describe con este detalle la herida con una flecha envenenada: “… porque la herida mortal hace que el lugar traspasado soporte el más insufrible tormento del mundo y aguarde la más fea y lamentable muerte; algunas veces mueren rematadamente locos, otras los intestinos se salen del vientre, y tan hediondos que ningún hombre puede soportar curarlos o atenderlos…”6. Obviamente nada de lo que refiere el corsario se compadece con lo que hoy podemos saber de una “curarización” ni de lo que vieron aquellos que después de Raleigh ensayaron el curare en forma experimental.
Era tal el temor a estas historias que se creó la necesidad de aprender sobre la naturaleza de este veneno, por lo que debía ser llevado a Europa y estudiado científicamente. Es así como los primeros experimentos llevados a cabo sobre este llamado ourara, urali, urare, woorari, wourali y curare (intentos europeos para reproducir la palabra nativa), fueron realizados en la Universidad de Leiden, Holanda, alrededor de 1740. Allí, un inglés de apellido Brockelsby, observó que incluso al ser inyectado el veneno en la pata de un gato, la respiración parecía comprometerse, pero el corazón seguía latiendo por más de dos horas, lo que significaba que el veneno no producía un paro cardíaco. Sin embargo, hacia 1780, el abate florentino Félix Fontana, en la misma universidad, observó su inocuidad por vía oral en el ser humano (a diferencia de lo que pasaba con cobayos y palomas), su incapacidad para matar a las víboras y, definitivamente, que sus vapores no causaban daño alguno4. Así, las historias difundidas pos los primeros navegantes eran erradas. Sólo estudios realizados en Israel en el Instituto Weizman más de dos siglos después, lograron demostrar que una de las subunidades que compone el receptor de acetilcolina en las víboras, tenía una secuencia diferente en la cadena de aminoácidos que la conforma (la secuencia entre el aminoácido 128 y 142)7. Esto hace que el curare y el veneno de las propias víboras no las afecten y que la respuesta en los animales sea diversa.
El investigador y explorador Alexander von Humboldt también estuvo involucrado en la historia del curare. Después de una serie de exploraciones hacia el interior de Venezuela realizadas entre 1799 y 1804, en 1832 dio la primera versión en occidente de la forma en que era preparado el veneno en Esmeralda, “el establecimiento cristiano más aislado y remoto del Alto Orinoco”8. Humboldt describe que el curare sería un veneno al ser usado en las puntas de flechas y dardos, pero también un medicamento al ser utilizado por vía oral. Probablemente la descripción más antigua del uso del curare en medicina fue hecha por un amigo y compañero de exploración de Humboldt, Robert Schomburgk, que probó el efecto “placebo” del curare en un ataque de malaria. Había visto utilizarlo a los nativos para los dolores abdominales pero obviamente era inefectivo para lo uno y para lo otro.
La siguiente colaboración en descubrir la causa de muerte del curare, fue hecha por Charles Waterton, un excéntrico viajero naturalista y taxidermista que, en su primer viaje a lo que después sería la Guyana Británica, en 1812 a la edad de 30 años, observó los efectos letales de dardos y flechas envenenadas y consiguió muestras del veneno para llevarlas a Europa 9 . Si Waterton no hubiera llevado muestras del veneno a Inglaterra, la historia del curare se hubiera detenido por algún tiempo. Con estas muestras hizo un experimento inyectando una dosis en la pierna de un asno después de efectuar una firme ligadura por sobre el sitio de punción, observando que el animal caminaba y se alimentaba normalmente; después de una hora, soltó la ligadura y en diez minutos el animal se desplomó y murió.
Benjamin Brodie fue un distinguido cirujano que estudió los efectos de una serie de venenos de origen vegetal y publicó sus observaciones en “The Effects of Certain Vegetable Poisons”. Su versión es que habría obtenido las muestras de “wourali” del Dr. Edward Nathaniel Bancroft, hijo de un médico que había ejercido en Guyana. De una personalidad muy diferente a Waterton y una rigurosidad científica incuestionable, llegó a ser presidente de la Royal Society. Después de ensayar y publicar experimentos en conejillos de India, gatos y conejos, hizo una demostración similar a la de Waterton en 1812 en la Royal Society en Londres, esta vez en un asno hembra, pero sin ligar la extremidad. En esta oportunidad el veneno, que ahora llamó “woorari”, paralizó rápidamente al animal y detuvo su respiración. Brodie practicó inmediatamente una traqueotomía y resucitó al animal envenenado manteniendo una ventilación rítmica durante dos horas. La burra sobrevivió varios años después del experimento. Es así como Brodie demostró finalmente como el curare mataba a sus víctimas: causando parálisis de la musculatura respiratoria, lo que resultaba en muerte por asfixia, sin ningún efecto sobre el cerebro o el corazón.
Uno de los problemas para tratar de averiguar qué era realmente el curare, es que las muestras llevadas a Europa tenían diferencias en variedad y cantidad de ingredientes, algunos de los cuales podrían deteriorarse con el tiempo. Lógicamente el veneno variaba en su composición de acuerdo a la región en que fue producido. Aquéllos obtenidos en la cuenca del Orinoco se componían principalmente de cortezas y raíces de la enredadera Strychnos nux y le llamaron Strychnos toxifera. Puesto que contenían estricnina, predominaban los efectos convulsivantes, enmascarando el efecto paralizante del curare. Por el contrario, aquéllos preparados en la jungla más occidental de lo que hoy es Ecuador y Perú, en la cuenca del río Marañón, se componían de un extracto de piel de vicuñas y especialmente de la enredadera Chondrodendron tomentosum. Esta última contenía altas concentraciones de curare y era similar a la utilizada por Waterton y Brodie en sus experimentos.
Así, se venía el enigma de descubrir como y porqué el curare producía parálisis, lo que fue realizado por los elegantes experimentos realizados por Claude Bernard en Francia, la intuición de Otto Loewi en Viena y los estudios de Sir Henry Dale en Londres.
Claude Bernard, Otto Loewi y Henry Dale
Los experimentos de Claude Bernard (Figura 2), fundamentales en la historia del curare, aunque mucho más significativos en otros campos (el rol del hígado y el páncreas en la regulación de los niveles de glucosa), relacionaron las observaciones de Brodie y Waterton y el análisis de la forma en que actúa sobre el músculo mismo. Demostraron la forma en que el curare causa la parálisis de la musculatura respiratoria y del aparato locomotor, y de este modo la muerte del animal. Esto es, demostró donde actúa el curare para paralizar a sus víctimas, mediante un meticuloso método científico.

Figura 2. Claude Bernard (1813-1858), fisiólogo francés cuyos experimentos realizados en preparaciones de músculo y nervio en diafragma de sapos fueron centrales en la historia del curare.
Un colega de Bernard, Jules Pelouze, llevó al laboratorio muestras de curare, traídas desde Brasil. Los resultados de los experimentos realizados por Claude Bernard con estas muestras de curare efectuados entre 1844 y 1856 fueron los que revelaron la forma en que el curare paralizaba y mataba a sus víctimas.
Como resultado de una serie de ensayos en músculos y nervios aislados de rana, Bernard logró demostrar que el curare envenenaba la conducción nerviosa pero no la contractilidad muscular. De este modo, estaba convencido que el curare prevenía de alguna manera los mensajes llevados por los nervios hacia la musculatura para producir contracción muscular. Es así que después de otra serie de elegantes experimentos para discriminar si el curare actuaba sobre nervios motores y sensitivos o selectivamente, logró demostrar que el curare actuaba sólo bloqueando los mensajes hacia los nervios motores que llegaban a los músculos, y concluyó que selectivamente y reversiblemente envenenaba a los nervios. Sus observaciones fueron publicadas en el famoso libro Leçons sur les effects des substances toxiques et medicamenteuses en 185610. Estaba casi en lo cierto.
Contemporáneamente, el desarrollo de microscopios de gran resolución en Alemania, permitió que Virchow demostrara que los órganos estaban formados por células independientes, lográndose el desarrollo de la histología. Así, en 1862, Wilhelm Kühne logró demostrar una leve hinchazón en el extremo del nervio motor, que terminaba es una especie de “bulbo”, y que había un espacio que lo separaba del músculo. También observó que la superficie opuesta, era diferente al resto del músculo, descubriendo de este modo la placa motora o el sitio de unión neuromuscular.
En 1866, un pupilo de Claude Bernard, Alfred Vulpian, concluyó en su tesis que este era probablemente el sitio de acción del curare. Reportó que cuando éste era aplicado cerca de la placa motora producía una parálisis rápida y profunda, mientras que, si se aplicaba cerca del nervio motor, producía un efecto menor y más lento. Concluyó así que “el curare interrumpe la comunicación entre las fibras nerviosas y las musculares”. En este caso, estaba completamente en lo cierto, aunque fue refutado por Bernard. No siempre los maestros reconocen las capacidades de sus discípulos, o peor aún, algunos sienten envidia de sus logros.
Con la invención del galvanómetro y el uso de largos nervios, como el del calamar gigante, se logró demostrar que una onda de corriente eléctrica pasaba a lo largo del nervio cuando era activado. Al alcanzar el término del nervio, la corriente causaba una respuesta, como la contracción de un músculo o la secreción de una glándula. La teoría eléctrica, que postulaba que la conducción de un estímulo nerviosos al músculo era el resultado de una carga eléctrica que saltaba el espacio entre la terminación nerviosa y la fibra muscular no fue cuestionada hasta los años 20 del siglo pasado. El afamado profesor de fisiología Charles Scott Sherrington era el mayor defensor de esta equivocada teoría. Aquéllos que postulaban la posibilidad de que estuviera involucrado un transmisor químico, eran considerados disidentes.
Fue sólo entre los años 1934 y 1950, que finalmente la opinión de los pocos disidentes a la teoría eléctrica fue tenida en cuenta y demostrada, con mucha paciencia y perseverancia. Como resultado de este terremoto científico, con epicentro en Cambridge, el paisaje de la fisiología humana fue definitivamente alterado. Durante ese período se hicieron los experimentos y se publicó la evidencia que establecía definitivamente que un agente químico, la acetilcolina, estaba involucrada en el traspaso de la información desde el cerebro, a través de los nervios, hacia los órganos bajo su control, entre ellos, la musculatura estriada.
Hay un acuerdo general en que el gran adelanto que produjo el cambio de paradigma entre la teoría eléctrica y la posibilidad de una transmisión química se debió a Otto Loewi y Henry Dale. Su contribución fue reconocida en 1936, cuando juntos recibieron el Premio Nobel por su trabajo sobre la transmisión química de los impulsos nerviosos.
Otto Loewi (Figura 3) estuvo en Cambridge, y bajo la influencia de Dale, avocado al estudio de la transmisión química de la transmisión neuromuscular, se dedicó al estudio del sistema nervioso autonómico a su regreso a Austria. Mediante una serie de prolijos experimentos llevados a cabo en corazones aislados de sapos (con su inervación conservada), logró demostrar por primera vez que un agente químico (la acetilcolina), estaba involucrado en pasar la información que era llevada por un nervio. Su valioso trabajo fue interrumpido al ser arrestado por los nazis, que ocuparon Austria en 1938. Estaba tan preocupado de que sus descubrimientos se perdieran que, desde la prisión, sobornando a un asistente, logró que sus estudios fueran enviados a una revista científica11. Posteriormente, gracias a los esfuerzos de la comunidad científica internacional y especialmente de Henry Dale, fue obligado entregar los ingresos de su Premio Nobel a los nazis y liberado. Posteriormente pudo seguir su actividad docente en los Estados Unidos.

Figura 3. Otto Loewi (1873-1961), fisiólogo alemán de origen judío que logró demostrar que el impulso nervioso se transmitía en forma química y no eléctrica, y que en el sistema nervioso simpático era la acetilcolina.
Cuando Henry Dale (Figura 4) pudo demostrar a la comunidad científica que se producía acetilcolina después de estimular un nervio, en ausencia de cualquier actividad muscular, fue cuando el curare ocupó un lugar protagónico. Al estimular el nervio que inerva el muslo de un animal, se producía acetilcolina tanto si el curare estaba presente o no; esta evidencia, conjuntamente con el hecho de que la acetilcolina inyectada en el vaso que irriga el músculo producía contracción muscular, aun si el nervio no era estimulado, no sólo probó la teoría de la transmisión química sino demostró la forma en que actuaba el curare. El curare había prevenido que la acetilcolina liberada durante la estimulación nerviosa, causara contracción del músculo: había “bloqueado” la acción de la acetilcolina en un área especialmente sensible del músculo12.

Figura 4. Henry Dale (1875-1978), fisiólogo inglés que aisló laacetilcolina, lo que permitió, junto a las investigaciones de Otto Loewi, demostrar la transmisión química del impulso nervioso. Ambos compartieron el Premio Nobel de Fisiología-Medicina en el año 1936.
Posteriormente, dos fisiólogos del Instituto Max Planck en Alemania, Neher y Sackman, finalmente demostraron como trabajan los transmisores químicos a nivel molecular y como su acción era bloqueada por el curare. También fueron recompensados con el Premio Nobel en 1991. Aislando un receptor de acetilcolina del músculo con microscopía electrónica, lograron demostrar que la acetilcolina abría el poro central en un área especial de la placa motora, ahora llamado receptor, lo que permitía que iones de sodio (eléctricamente cargados), pasaran a través de la membrana celular, lo que se traducía en la generación de un micro impulso eléctrico. La producción de una pequeña corriente eléctrica era entonces el efecto, y no la causa, de la transmisión neuromuscular. Además, este efecto era bloqueado por el curare 13 .
Uso clínico del curare
Muy precozmente, después de los experimentos de Bernard, en forma bastante empírica se usó curare en enfermedades que producían contractura muscular, como tétanos y rabia, y en enfermedades convulsivas como la epilepsia. Se publicaron muchas descripciones sobre el uso de curare para el tratamiento de convulsiones alrededor de 1950, y muchos se atribuyeron el crédito de haber logrado una terapia. Obviamente los resultados no fueron halagadores porque, si bien los pacientes disminuían su contractura, los síntomas se manifestaban nuevamente al pasar el efecto, o incluso algunos caían en depresión respiratoria y muerte. La alternativa de entonces eran dosis crecientes de láudano (opio), que también mejoraba los síntomas, pero hacía caer a los pacientes en depresión respiratoria y anoxia. No habían llegado aún los tiempos de la ventilación artificial.
En la introducción del curare en clínica no podía estar ausente la serendipia. Richard Gill, ex director de una compañía de caucho, decidió instalarse al oriente de la cordillera de los Andes, en la cuenca amazónica de Ecuador. Tuvo un accidente al caer de su caballo que le dejó con una paresia espástica, probablemente una lesión medular, que lo hizo volver a Estados Unidos para recuperarse lentamente con rehabilitación. Durante su recuperación sufrió severos espasmos dolorosos en las extremidades derechas. Fue entonces cuando pensó en la posible utilidad del veneno usado por los nativos vecinos a su finca en Sudamérica para cazar a sus presas. Esto lo hizo retornar a Ecuador en 1938 para recolectar suficiente curare para que la droga pudiera ser purificada y ensayada clínicamente en Estados Unidos. Así, logro obtener unas 39 libras de curare crudo (unos 13 kilos) 14 . De este modo, si Gill no se hubiera caído del caballo, la historia de la medicina no hubiera contado tan rápidamente con este esencial producto, pues hasta entonces, el curare no era más que una curiosidad farmacológica. Por supuesto Gill nunca se recuperó totalmente, permaneciendo con episodios de espasticidad y temblor. Su uso para aliviar la espasticidad no tuvo éxito, pero fue la serendipia la que produjo la convergencia de eventos que condujeron a la llegada de esta gran cantidad de droga a los Estados Unidos, y que permitió los ensayos clínicos en el campo de la anestesiología 3 .
Fue en los laboratorios de E. R. Squibb que el curare fue purificado y estandarizado por primera vez en una preparación comercial. Sin embargo, fue Harold King en 1935 quien aislara e identificara por primera vez la estructura química del curare en los Laboratorios Burroughs Wellcome de Londres a partir del Condrodendron y puesto que la muestra había sido guardada y rotulada “Ucayalli river 1871” en un recipiente de bambú, al alcaloide purificado se le llamó tubo-curarina15.
King demostró que la estructura química del curare era muy similar a dos moléculas de acetilcolina, separadas por un enlace químico voluminoso. Tenía la forma de dos ganchos, separados por un vástago; la distancia entre ambos extremos tenía que tener una dimensión específica. Esto permitía que los extremos de la molécula engancharan simultáneamente como llave en una cerradura en el receptor, bloqueando la transmisión neuromuscular. Si la distancia entre las dos moléculas varía, sólo un gancho se adhiere a la vez y no se produce bloqueo. Aunque ahora sabemos que la fórmula de King no era completamente exacta, tuvo el mérito de iniciar la síntesis de nuevas moléculas de curare, más selectivas en su acción bloqueadora neuromuscular y con menos efectos colaterales. Es lo que posteriormente se llamó relación estructura-actividad.
El primer curare purificado por laboratorios Squibb fue llamado Incostrin y fue usado curiosamente en Nebraska, para disminuir las convulsiones asociadas a la terapia convulsiva con Metrazol, una droga usada en siquiatría como alternativa al electroshock. El siquiatra, de apellido Bennett, reportó que los pacientes dejaron de tener fracturas por la convulsión, utilizando “la mitad de la dosis necesaria de Incostrin que producía parálisis respiratoria”. Sin embargo, la introducción en anestesia propiamente tal pertenece al entusiasta y persistente Lewis Wright.
Wright, formado como anestesiólogo en el Bellevue Hospital de Nueva York, fue capaz de observar la dificultad asociada a producir buenas condiciones quirúrgicas al abrir el abdomen y relacionarla con el uso del curare, que había observado usar a Bennett en la prevención de convulsiones. Su bagaje anestesiológico lo hizo pensar en administrar más relajación y menos anestesia en los pacientes más enfermos, ya que la forma de hacerlo era hasta entonces profundizando la anestesia. Financiado por el laboratorio Squibb, intentó convencer a los líderes de opinión en anestesiología de los Estados Unidos de entonces en el Congresos de Nueva York en 1940. Convenció a su antiguo jefe en el Bellevue, el Dr. Emery Andrew Rovenstein, quién usando la misma dosis sugerida por Bennett, terminó con el paciente en paro respiratorio, requiriéndose respiración artificial (algo no muy común en 1949), hasta que lentamente recuperó la ventilación. Se repitió el experimento con menos dosis con similares resultados, por lo que Rovenstein decidió abandonar, por considerar que la droga excedía el margen de seguridad. Una experiencia similar tuvo con el Dr. Stuart Cullen, uno de los pocos anestesiólogos de entonces en Estados Unidos con experiencia en investigación. Cullen ensayó la droga en perros con fatales resultados. El fracaso con Rovenstein se produjo por la potenciación del efecto relajante del curare con el éter. En aquellos tiempos, la depresión respiratoria se asociaba a exceso de anestesia y los pacientes no eran intubados. El fracaso de Cullen, por una gran diferencia interespecie del curare, el que en perros produce una liberación de histamina incompatible con su uso clínico.
Pero Wright no estaba dispuesto a abandonar. En otro encuentro de anestesistas, sin darse por vencido, contactó al Dr. Harold Randall Griffith, jefe del Departamento de Anestesiología del Homeopatic Hospital en Montreal (futuro Queen Elizabeth Hospital), formado en Inglaterra y responsable de haber incorporado recientemente el ciclopropano en Canadá. A diferencia de los anestesiólogos americanos, Griffith solía usar la intubación endotraqueal, para evitar la depresión respiratoria, asistiéndolos manualmente. Finalmente, Griffith aceptó probar el Incostrin, aunque con cierto recelo, y con mucha preparación por si ocurriera algún incidente. El primer ensayo tuvo lugar el 22 de enero de 1942, esta vez usando ciclopropano, que no produce el efecto potenciador del bloqueo del éter. Es así como, conjuntamente con su colega Enid Johnson, Griffith utilizó Incostrin en 25 pacientes sin ninguna complicación, publicando sus resultados en el American Journal of Anesthesiology en 194216, abriendo de este modo una nueva era en la anestesia. Prontamente, el hecho fue reconocido en Europa como “un hito en la anestesia”. Se había cambiado un paradigma y se había abierto un nuevo capítulo en la práctica de la anestesia.
Siguieron otras publicaciones en los Estados Unidos 17 , donde siempre se usó el Incostrin con precaución por la falta de preparación de los anestesiólogos y el ejercicio de la especialidad por enfermeras no aptas para el manejo de la vía aérea. Después de la guerra, en 1946, los Drs. Cecil Grey y Jack Halton usaron d-tubocurarina -el curare sintético que había preparado el laboratorio Burroughs Wellcome- en 1.000 pacientes en Liverpool, presentándolo en la Royal Society of Medicine, en Londres, con el el título: “A Milestone in Anaesthesia?” 18 . Así comenzó su uso generalizado en Europa, donde los anestesistas tenían una preparación médica y un adecuado manejo de la vía aérea, permitiendo un bloqueo neuromuscular total, manejado con ventilación artificial.
Ahora bien, si se quiere ser apegado a los hechos y no a la historia más difundida, el primero en estudiar el curare en experimentación animal y administrarlo en una anestesia general fue Arthur Läwen, un cirujano de Leipzig en 1912. Aunque el resultado de un estudio realizado en 7 pacientes sometidos a anestesia general para facilitar el cierre de la pared abdominal fue publicado, Läwen no pudo seguir usándolo por falta de suministro y su contribución a la historia de la anestesia ha sido casi siempre olvidada 19 .
En relación al uso clínico del curare se desarrollaron dos escuelas. En Estados Unidos, probablemente inducidos por las primeras experiencias, lo consideraban inherentemente peligroso. Además, la práctica de la anestesia era realizada mayormente por no especialistas. De este modo, se recomendaban sólo pequeñas dosis de curare, como suplemento de la anestesia general. En Inglaterra y el resto de Europa, la aproximación al uso del curare era diferente, usándose dosis totalmente paralizantes, con control de la ventilación y una anestesia más superficial. Inglaterra era el único país donde la anestesia era administrada casi exclusivamente por médicos calificados y posteriormente anestesiólogos, con reputación científica desde los tiempos de John Snow. El resultado fue que los estándares de la práctica anestésica y la investigación fueron los mejores del mundo.
En la actualidad, especialmente con el desarrollo de drogas con gran margen de seguridad, la técnica inglesa prevaleció, de modo que la mayoría de los pacientes reciben una dosis paralizante de BNM, se controla la ventilación y se administra un antagonista al término del procedimiento. Con esta técnica, debe tenerse especial cuidado en que la relajación muscular no oculte una anestesia superficial.
Después de 25 años de uso de curare en Inglaterra, un estudio presentado en el Royal College of Surgeons reveló una disminución de la mortalidad de causa anestésica de un 30%, cuya causa en gran parte podía atribuirse el uso de BNM y un mejor entendimiento de la fisiología relacionada con su uso 3 . Sin embargo, un estudio realizado en 1954 por Beecher y Todd20, que recibió bastante publicidad, mostró que, por el contrario, el uso de curare sextuplicaba el número de complicaciones postoperatorias y muertes. El estudio cubría un período de 4 años entre 1948 y 1952 en un total de 600.000 anestesias. Se publicaron así una serie de artículos contradictorios que se refutaban unos con otros, pero la conclusión del famoso artículo de Harvard fue pronto modificada. El aumento de la mortalidad se había debido a la aceptación de enfermos cada vez más comprometidos para la cirugía y a pobres estándares de seguridad del manejo anestésico en Estados Unidos, en que se usaba parálisis parcial, con mal manejo de la vía aérea, por personal paramédico o médico mal entrenado y no se usaba reversión del bloqueo residual.
La Historia Moderna
Hasta 1967, el curare era el BNM disponible más asequible en nuestra especialidad y su margen de seguridad cardiovascular era insuficiente. Con el desarrollo de la cirugía coronaria, la hipotensión que causaba la liberación de histamina empezó a constituir un problema y se trató de encontrar drogas con un perfil más seguro, tanto desde el punto de vista hemodinámico como metabólico.
En 1946, Daniel Bovet descubriría la galamina, un compuesto con tres nitrógenos cuaternarios, que sería introducido por Huguenard y Boué en 1948 y que presentaba algunas ventajas sobre la d-tubocurarina 21 . Fue el primer BNM sintético usado en clínica, pero su efecto extremadamente vagolítico y exclusiva eliminación renal lo hicieron pronto impopular, especialmente después de la aparición del pancuronio. Nunca se usó en Chile.
Por azar, el mismo año 1945 dos grupos independientes, Barlow e Ing, y Paton y Zaminis, sintetizan independientemente el decametonio, que fue introducido en clínica en 1949 por G. S. W. Organe 22 . Así aparece por primera vez el grupo de los metonios en la historia de los BNM, demostrando una gran diferencia de actividad bloqueadora neuromuscular o bloqueadora ganglionar según el largo de la cadena entre 2 amonios; entre ellos, el suxametonio (o succinilcolina en Estados Unidos), cuya calidad de bloqueo y tiempo de inicio de acción pasó a formar parte del listado de características ideales.
En 1906 Hunt y Taveau habían descrito los efectos de la succinilcolina y una serie de análogos de la colina sobre la presión arterial en gatos, pero nunca advirtieron su efecto relajante muscular, pues los ensayos se hicieron en animales curarizados 23 . Es así que su efecto bloqueador neuromuscular fue descubierto sólo después de 43 años por Bovet y Phillips 24 . Finalmente fue introducida en la práctica clínica en Europa por Brücke en Viena en 1951 25 , Thesleff en 1951 en Estocolmo26 y en Estados Unidos por Foldes en 1952 27 . Fue justamente la descripción de Foldes en su artículo la respuesta del por qué de su vigencia: “Comparada con otros relajantes, la succinilcolina posee varias ventajas, siendo la más relevante su fácil control, que permite cambios casi instantáneos en el grado de relajación muscular. Con la succinilcolina, tanto el aumento como la disminución de la relajación muscular, toman menos de un minuto.” El rápido comienzo del efecto relajante, su rápida reversión y su precio económico son factores que probablemente han favorecido la permanencia de su utilización a pesar de sus efectos secundarios.
En 1957 el Profesor Daniel Bovet recibió el Premio Nobel de medicina por sus estudios relacionados con los BNM galamina y succinilcolina, que fueron (y la succinilcolina sigue siendo), de gran utilidad en anestesia. El crédito de Bovet es indudable, pero la síntesis de ambas drogas fue hecha por Ernest Foumeau y Reid Hunt respectivamente; desgraciadamente, estos dos científicos han sido en gran medida ignorados 28 .
Dos conceptos falsos guiarían la búsqueda de nuevos BNM por varios años: que era indispensable la presencia de dos nitrógenos cuaternarios con una distancia de 1,2 a 1,4 nm entre ellos y que la incorporación de ellos en una estructura rígida de anillos heterocíclicos facilitaba el bloqueo.
La revisión de la estructura definitiva de la d-tubocurarina hecha por Everett, cambió la línea de investigación 29 . A partir de entonces se aceptó que el bloqueo neuromuscular podía ocurrir con moléculas con un solo nitrógeno cuaternario, y que la distancia entre ellos tenía menos importancia. Adquirieron importancia factores como la estereoquímica, la presencia de grupos funcionales que favorecieran el metabolismo o la degradación de la molécula, el grado de hidro o lipofilicidad que pudieran modificar la farmacocinética (distribución y eliminación) y el diseño molecular para disminuir o suprimir los efectos secundarios. El esfuerzo se concentró en buscar un agente considerado como “BNM ideal”, desarrollándose dos líneas de investigación, que han entregado al arsenal anestesiológico los agentes más utilizados en la actualidad. Es así como a partir de productos naturales o totalmente sintéticos, se desarrollaron alrededor de 45 fármacos, todos ellos derivados sólo de dos grupos químicos: los esteroides y las bencilisoquinolinas 30,31 .
Sólo quedaron fuera de estos grupos, que vale la pena mencionar si se está hablando de historia, la c-toxiferina I y la dialil-nor-toxiferina (alcuronio), cuya molécula híbrida sugiere una estructura esteroidal por su nombre, pero que es más bien un curare. Tuvo un efímero pasar, con un rápido reemplazo cuando se comercializó el pancuronio, debido a su gran efecto ganglioplégico 32 .
Aminoesteroides
El pancuronio fue el primer BNM de síntesis con estructura aminoesteroidal y propiedades bloqueadoras neuromusculares usado en clínica 33 . La historia comienza en los 60 a partir de la síntesis de la maluetina, un alcaloide esteroidal aislado de la corteza del arbusto Malouetia bequaertiana, que producía bloqueo neuromuscular. Después de 4 años de investigación Hewett y Savage sintetizaron un aminoesteroide con dos grupos de amonio cuaternario, y en un notable trabajo de ingeniería química, unieron dos fragmentos similares a la acetilcolina a un anillo esteroide rígido de androstano de 17 átomos de carbono, para conseguir la acción específica de bloqueo neuromuscular (Figura 5) 34,35 . Fue introducido en clínica en 1967 por Baird y Reid36. Aunque similar a la d-tubocurarina en cuanto a su inicio y duración de acción, su margen de seguridad cardiovascular era mucho mejor, sin presentar liberación de histamina y bloqueo ganglionar, aunque bloqueaba los receptoress muscarínicos cardíacos, produciendo un moderado efecto vagolítico.
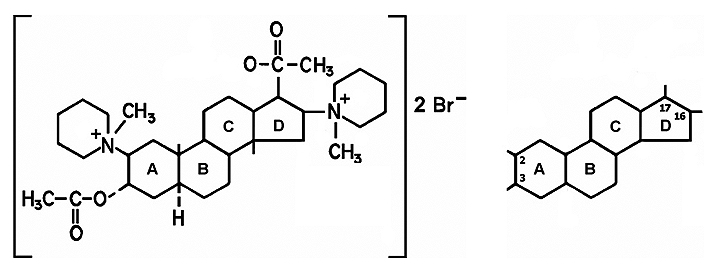
Figura 5. Pancuronio (izquierda): fragmentos similares a la acetilcolina, unidos a un anillo esteroide rígido de androstano (derecha), a partir del cual se sintetizaron los compuestos de estructura similar, por substituciones en las posiciones 2, 3, 16 y 17.
El primer paso por mejorar el pancuronio ocurrió en Hungría, donde la síntesis del pipecuronio, que difiere del pancuronio en las substituciones 2 y 16 del núcleo androstano, dio como resultado un relajante muscular de larga duración, pero sin efecto vagolítico. Este agente, mucho mejor que su precedente, no se usó en occidente sino hasta muchos años después, como resultado de la falta de intercambio comercial durante la Guerra Fría37.
A partir del anillo básico del androstano, y mediante una serie de substituciones, especialmente a nivel de los átomos de carbono 2, 3 y 16, 17, se sintetizaron todos los otros compuestos de estructura esteroidal (vecuronio, pipecuronio, rocuronio y rapacuronio). Todos tienen dos átomos de nitrógeno cuaternario, situados a una distancia de 11,1 Å38. La Figura 6, muestra el núcleo androstano, con las diferentes substituciones que han dado origen a los BNM del grupo aminoesteroide. Pueden identificarse dos series de compuestos monocuaternarios estrechamente relacionados, de los que se comercializaron el vecuronio, el rocuronio y el rapacuronio, que pronto fue sacado del mercado. Una serie de compuestos 16-N-metil, variando sólo el sustituyente éster en la posición 17: vecuronio (17-acetil), Org 9489 (17 propionil) y Org 9453 (17-butiril). La otra serie está constituida por compuestos monocuaternarios 16-N-alil, variando principalmente, pero no exclusivamente, el sustituyente éster en la posición 17: rocuronio (17-acetil), rapacuronio (17-propionil) y Org 7617 (17-butiril). El rocuronio tiene también otros sustituyentes y una desacetilación en la posición 3, que lo hacen obtener estabilidad en solución acuosa.
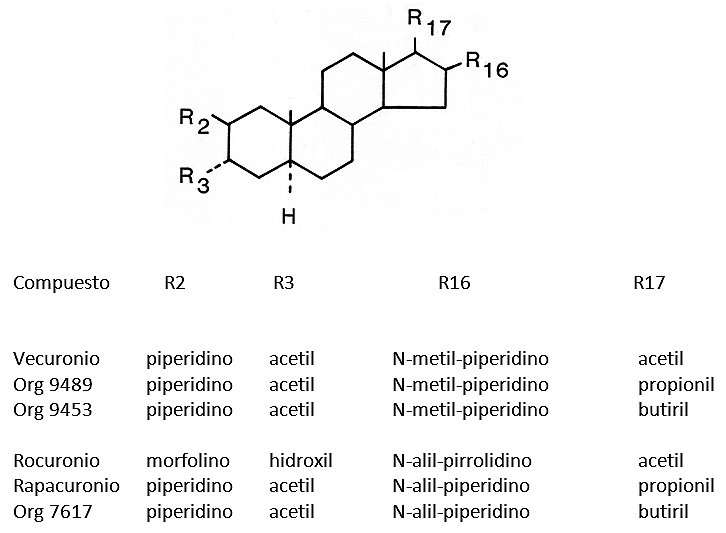
Figura 6. Estructura molecular de una serie de relajantes musculares del grupo aminoesteroide sintetizados por Organon Laboratories, tres de los cuales han sido comercializados.
La atención se centró ahora en la búsqueda de un BNM que fue llamado “ideal”, de menor duración y menor tiempo de inicio de acción 39 . Así empieza la carrera por investigar simultáneamente la búsqueda de un compuesto de duración intermedia, con el menor efecto cardiovascular posible, siendo el resultado la incorporación de dos drogas que convivieron por casi veinte años: el vecuronio, en el grupo de los aminoesteroides, y el atracurio en el grupo de las bencilisoquinolinas. Luego, intentando obtener un agente no despolarizante con un tiempo de inicio similar a la succinilcolina, se logró la síntesis del rocuronio y el rapacuronio.
Savage pudo reconocer que la actividad estimuladora de los receptores muscarínicos cardíacos que producía taquicardia con el pancuronio, residía en un uno de los amonios cuaternarios del extremo del núcleo androstano (en el anillo D) y el efecto bloqueador neuromuscular en el nitrógeno cuaternario del otro extremo (en el anillo A). Así, desmetilando el nitrógeno cuaternario del anillo A, logró sintetizar el vecuronio, un agente de duración de acción intermedia (ni tan larga como el pancuronio, ni tan corta como la succinilcolina), pero desprovista de todo efecto cardiovascular. El vecuronio fue introducido en clínica en 1980 40, 41 .
El siguiente gran avance en el desarrollo de los BNM del grupo aminoesteroide se produjo al partir del trabajo de Bowman, que en 1988 estableció que en los BNM de este grupo químico la velocidad a la que ocurría el bloqueo (tiempo de inicio de acción) era proporcional a la potencia del agente. Es decir, los fármacos con menos potencia (aquellos que requerían más dosis para producir el mismo efecto), tenían un tiempo de inicio de acción más rápido 42 . Así surgieron el rocuronio y el rapacuronio. Se hicieron varias modificaciones químicas a la molécula de vecuronio, llegándose al bromuro de rocuronio, que difiere estructuralmente del vecuronio en 4 posiciones del núcleo esteroidal: tiene un grupo 2-morfolino, un grupo 3-hidroxi y una función 16-pirrolidino unida a un grupo 16-N-alilo 43 . El rocuronio tiene un perfil muy similar al vecuronio, pero en dosis adecuadas logra un tiempo de inicio de acción muy similar al de la succinilcolina. Fue introducido en clínica en Estados Unidos en 1995 y en Chile en 1996 44 .
El último aporte al arsenal farmacológico del los BNM del grupo aminoesteroide, digno de ser mencionado históricamente fue el rapacuronio 45 . Es el análogo 16N-alil-17-propionato del vecuronio. Era el primer BNM no despolarizante que combinaba las atractivas características de rápido inicio de acción y corta a intermedia duración de acción 46 . Llamó la atención la rápida aprobación hecha por la FDA en 1999, cuando la droga recién comenzaba la fase clínica de investigación, cuando había pocos estudios en animales y muy pocos estudios en seres humanos publicados en la literatura médica. Pronto, dos años después de su introducción en clínica, se reportaron broncoespasmos severos, que hicieron al laboratorio fabricante retirarlo rápidamente del mercado 47 .
Bencilisoquinolinas
Poco después de la publicación de la estructura correcta de la d-tubocurarina realizada por Everett en 1970, casualmente, John B. Stelanke, investigando un alcaloide de los tubérculos de una planta mediterránea, Leontice leontopetalum, sugirió una aproximación completamente novedosa y desconocida en el metabolismo de los mamíferos. Uno de los principales constituyentes de dicha planta, la petalina, que era una simple sal bencilisoquinolínica cuaternaria, tenía algunas similitudes estructurales con la d-tubocurarina y, además, sufría la apertura de un anillo y conversión a una amina terciaria de cadena abierta en la presencia de un suave medio alcalino.
Se trataba de un antiguo proceso que había sido denominado por los químicos “eliminación de Hof-mann”, en honor a A.W. Hofmann que, en 1851, en el mismo año en que Claude Bernard concluía sus experimentos con el curare, había descrito que a 100°C, las sales de amonio cuaternario pueden descomponerse en un medio fuertemente alcalino para formar una base terciaria. Más de 120 años después, Stenlake, al observar que no era indispensable un fuerte medio alcalino y una alta temperatura para que ocurriera este proceso, vislumbró la posibilidad de sintetizar un BNM que pudiera ser autodestruido en el organismo, activado por el leve medio alcalino del pH fisiológico y sin la intervención de mecanismos hepáticos o renales.
Esta observación dio origen a una serie de estudios cuyo objetivo era la síntesis, en 1981, de un compuesto bicuaternario con características estructurales que permitieran un bloqueo neuromuscular competitivo y que sufriese una fácil degradación en condiciones fisiológicas de pH 48 . Se obtuvo así un fármaco de menor duración que los relajantes musculares usados entonces (d-tubocurarina, dialil-nor-toxiferina y pancuronio), más predecible en cuanto a potencia y duración del bloqueo y sin efectos acumulativos, cuyo principal atractivo ha sido su original vía de eliminación: el atracurio, introducido en clínica en 1984, igual que el vecuronio.
Investigaciones posteriores demostraron que el atracurio tiene al menos tres vías metabólicas importantes: la eliminación de Hofmann, la hidrólisis éster y la eliminación a través de los órganos, en la cual probablemente el hígado tiene la mayor participación. A lo largo de los años ha habido una gran especulación sobre la importancia relativa de cada una de estas vías. Los estudios más antiguos sugerían que la eliminación de Hofmann era la vía primordial, pero los ulteriores han demostrado que, en condiciones normales, la eliminación a través de los órganos es la más importante, llegando a un 60%. Más de la mitad del 40% restante es responsabilidad de hidrólisis éster y no de la eliminación de Hofmann, contrariamente a lo que se planteó en un comienzo 49 .
El atracurio y el vecuronio competirían por más de 20 años en el mercado de los BNM, y aquel sigue utilizándose por su característica única de independencia de eliminación del hígado y el riñón, cosa que nunca se ha logrado con los BNM del grupo aminoesteroide. Todo un mito circula en relación a la gran batalla librada por laboratorios competidores, para introducir un agente de duración intermedia, que finalmente ocurrió simultáneamente en 1984. De hecho, la investigación de ambos BNM se realizó bajo estricto secreto en instalaciones de laboratorios rivales, situados a unas escasas cuadras de distancia en Glasgow.
El atracurio es en realidad una mezcla racémica de 10 esteroisómeros, y produce algún grado de liberación de histamina. De estos 10 isómeros, se han ensayado 6 como BNM, pues los otros 4 son de síntesis muy difícil. Los 6 isómeros del atracurio estudiados varían su tiempo de inicio y su duración de acción en forma inversamente proporcional a la potencia del bloqueo 50 . Se seleccionó finalmente el isómero 1R cis-1’R cis, el único de los 6 que no libera histamina y que constituye normalmente el 15% de la mezcla racémica de atracurio, obteniéndose una droga más predecible desde el punto de vista farmacodinámico, el besilato de cisatracurio, incorporado por la FDA en los Estados Unidos en 1996, y disponible en Chile desde 1998 51 .
El grupo de Savaresee desarrolló también el mivacurio. Químicamente es un derivado bencilisoquinolínico, con un enlace éster y dos fragmentos similares a la colina; difiere del atracurio por la presencia de un grupo fenólico metilado adicional y en la posición relativa de los grupos ésteres. De este modo, es un BNM metabolizado por la colinesterasa plasmática, con un tiempo de inicio de acción similar a los BNM de duración intermedia, pero una duración clínica más corta que el de todos los BNM no despolarizantes. Fue introducido en clínica en 1988 52 .
Conclusiones
Al analizar la selección natural de lo BNM desarrollados desde la síntesis de la d-tubocurarina, puede constatarse 2 períodos:
Período 1942-1980
Durante el transcurso de este período se desarrollaron fundamentalmente productos procedentes de la modificación química de productos naturales (Chondodendrum spp, Strychnos spp, Erythrina corraloides), y solo secundariamente algunos compuestos puramente sintéticos. De la gran cantidad de BNM desarrollados en este período, muchos de ellos tuvieron una vida efímera, e incluso no se conocieron en Chile. Como dato a destacarse, durante todos estos años se han comercializado 15 productos, 8 de los cuales provenientes de tan sólo tres moléculas diferentes. En forma cronológica:
– 1942: d-tubocurarina
– 1946: Cloruro de d-tubocurarina
– 1948: Galamina
– 1947: Mefenesina
– 1949: Decametonio
– 1950: Metocurina
– 1951: Succinilcolina
– 1951: Benzoquinonio
– 1952: Laudexio
– 1955: Hexafluorenio
– 1959: Bromuro de hexametileno carbaminoicolin
– 1961: C-toxiferina I
– 1962: Dialil-nor-toxiferina
– 1967: Bromuro de pancuronio
– 1972: Fazadinium
La realidad práctica de estos primeros 40 años, es que habiéndose dejado de usar la mayoría de ellos (el fazadinium y el hexametileno en persistieron hasta 1990), el anestesiólogo se manejó fundamentalmente con:
No despolarizantes:
d-tubocurarina
Metocurina*
Gallamina*
Alcuronio
Pancuronio
Despolarizantes:
Succinilcolina
(* = no llegados a Chile).
Período 1980-2000
Durante este período se produjo el gran desarrollo de BNM de síntesis, provenientes de la substitución de diferentes radicales, en sólo dos grupos químicos: las bencilisoquinolinas, que habían tenido precursor en el benzoquinonio, y los aminoesteroides, que habían debutado con el pancuronio. Desde entonces, dos grandes conglomerados farmacéuticos compiten por el mercado de los BNM, y con la excepción del pipecuronio, que fue desarrollado en Europa del este y después absorbido por uno de ellos, todos los nuevos relajantes musculares se han sintetizado en sus laboratorios. En estos 20 años de investigación farmacológica, se pone a disposición de los anestesiólogos una serie de relajantes musculares que poseen una acción mucho más selectiva, menos efectos colaterales y gran versatilidad de indicaciones. En forma cronológica:
1980: Pipecuronio*
1984: Atracurio
1984: Vecuronio
1988: Doxacurio*
1994: Mivacurio
1995: Rocuronio
1996: Cisatracurio
2000: Rapacuronio*
(* = no disponibles en Chile).
La realidad práctica de estos últimos años ha sido una tendencia a la disminución del uso de BNM de larga duración. En Chile, está disponible sólo uno de la era 42-80 (la succinilcolina), y cinco de los nuevos o no tan nuevos (atracurio, vecuronio, mivacurio, rocuronio y cisatracurio). La succinilcolina tiene cada día menos uso en el adulto y salvo en el manejo de la vía aérea difícil, su uso está contraindicado en los niños. El pipecuronio, siendo la mejor droga de larga duración, dejó de comercializarse en Chile por su escasa salida, y el único relajante de este espectro disponible en Chile, el pancuronio, por su uso cada vez menor, tampoco está disponible en la actualidad. La tendencia ha sido que el rocuronio haya ido reemplazando al vecuronio y el cisatracurio no ha tenido el mismo proceso respecto del atracurio, debido especialmente a que su largo tiempo de inicio de acción es incompatible con la impaciencia de algunos anestesistas. El mivacurio es el único relajante muscular no despolarizante de espectro corta duración y tiene varias aplicaciones de uso clínico actual. El rapacuronio, como es por todos sabido, nació muerto.
Los BNM constituyeron un hito en la práctica de la anestesia clínica. A pesar de esto, todos los que están actualmente en uso tienen algunas limitaciones y la necesidad de un BNM “ideal” persiste, aunque la investigación en esta área ha disminuido porque se ha llegado a agentes con grandes márgenes de seguridad. Probablemente al BNM ideal de la actualidad se le pide bastante más que Savarese en los años 70. Hoy se hace necesario un agente de rápido inicio de acción (como la succinilcolina), que no se acumule, cuyo metabolismo sea independiente de la función hepática y renal (como el atracurio, el cisatracurio y el mivacurio), que sea fácilmente reversible (con un agente que no tenga efectos secundarios) y libre de efectos laterales, especialmente hemodinámicos (como el vecuronio y el cisatracurio). Ese híbrido no existe y no es parte de esta historia.
Conflicto de interés: ninguno.
Referencias
1. Utting JE. The era of relaxant anaesthesia [Editorial]. Br J Anaesth. 1992 Dec;69(6):551–3. https://doi.org/10.1093/bja/69.6.551 PMID:1467094
2. Raleigh SW. The discoverie of the large, rich and beautiful Empire of Guiana. London: R. Robinson; 1596.
3. Feldman S. From Poison Arrows to Prozac. Londres: John Blake Publishing; 2009.
4. Steinberg D, Chalita J. Versiones y controversias fascinantes sobre el curare. Relajantes musculares en Anestesia y Terapia Intensiva. Madrid: Álvarez JA, González F y Bustamante R. Segunda Edición. Arán Ediciones; 2000.
5. D’Anghiera PM. De Rebus Oceanis et Orbe Novo, 1516. Translation from latin by F. A. McNutt. New York: G. P. Putnam’s Son; 1912.
6. Raleigh W. Breve y admirable descripción del Reino de Guyana. Edimburgo; 1820.
7. Fuchs S. Molecular Evolution od the Binding Site of Acetilcholine Receptor. Ann N Y Acad Sci. 1994;•••:126.
8. Humboldt A. Maravillas y misterios de Venezuela. Diario de viajes 1799-1804. Versión de viajes a las regiones equinocciales del nuevo continente. Caracas: Editorial CEC; 1998.
9. Waterton Charles. Wanderings in South America, 1879. (Edición de 1839 de B. Fellowes, London, disponible en Proyecto Gutenberg).
10. Bernard C. Bailliere et fils. Leçons sur les effects des substances toxiques et medicamenteuses (Paris, tr. Thornton). Baltimore: Williams & Wilkin Press; 1961.
11. Loewi O. Über humorale übertragbarkeit der Herznervenwirkung. Pflugers Arch. 1921;189(1):239–42. https://doi.org/10.1007/BF01738910.
12. Dale H. Chemical transmission of the effects of nerve impulses. BMJ. 1934 May;1(3827):835–41. https://doi.org/10.1136/bmj.1.3827.835 PMID:20778253
13. Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976 Apr;260(5554):799–802. https://doi.org/10.1038/260799a0 PMID:1083489
14. Gill RC. White Water and Black Magic. New York: Henry Holy & Co.; 1940.
15. King H. Curare alkaloids: 1, tubocurarine. J Chem Soc. 1935;1381–9. https://doi.org/10.1039/jr9350001381 PMID:18914103
16. Griffith HR, Johnson GE. The use of curare in general anesthesia. Anesthesiology. 1942;3(4):418–20. https://doi.org/10.1097/00000542-194207000-00006.
17. Cullen SC. Curare; its rational use as an adjunct to anesthesia. Surgery. 1946 Aug;20:200. PMID:20994801
18. Gray TC, Halton J. A Milestone in Anaesthesia? (d-Tubocurarine chloride). Proc R Soc Med. 1946;39:400–10.
19. Goerig M, Schulte am Esch J. Arthur Läwen—Ein Wegbereiter moderner Anästhesieverfahren. Anasthesiol Intensivmed Notfallmed Schmerzther. 1993 Aug;28(5):315–25. https://doi.org/10.1055/s-2007-998932 PMID:8373979
20. Beecher HK, Todd DP. A study of the deaths associated with anesthesia and surgery: based on a study of 599, 548 anesthesias in ten institutions 1948-1952, inclusive. Ann Surg. 1954 Jul;140(1):2–35. https://doi.org/10.1097/00000658-195407000-00001 PMID:13159140
21. Wylie WD, Churchill-Davidson HC. A Practice of Anaesthesia. Segunda Edición. Chicago: Year Book; 1966.
22. Organe G, Paton WD, Zaimis EJ. Preliminary trials of bistrimethylammonium decane and pentane diiodide (C10 and C5) in man. Lancet. 1949 Jan;1(6540):21–3. https://doi.org/10.1016/S0140-6736(49)90345-1 PMID:18104889
23. Hunt R, Taveau R. On the physiological action of certain cholin derivatives and new methods for detecting cholin. BMJ. 1906;2:1788–91.
24. Bovet D, Bovet-Nitti F, Guarino S, Longo VG, Marotta M. Proprieta farmachodinamiche di alcuni derivati della succinilcolina dotati di azioni curarica. Rend Inst Sup Sanita (Roma). 1949;12:106.
25. Brücke H, Ginzel KH, Pfaffenschlager F, Werner G. Bis cholinester von dicarbonsaüren als muskelrelaxantien in der narkose. Wien Klin Wochenschr. 1951;63:464–572. PMID:14867297
26. Thesleff S. An investigation of the muscle-relaxing action of succinyl-choline-iodide in man. Acta Physiol Scand. 1952 Jun;25(4):348–67. https://doi.org/10.1111/j.1748-1716.1952.tb00886.x PMID:12976151
27. Foldes FF, McNall PG, Borrego-Hinojosa JM. Succinylcholine: a new approach to muscular relaxation in anesthesiology. N Engl J Med. 1952 Oct;247(16):596–600. https://doi.org/10.1056/NEJM195210162471603 PMID:12993276
28. Cozanitis DA. Daniel Bovet, Nobelist: muscle relaxants in anaesthesia : The role played by two neglected protagonists. Wien Med Wochenschr. 2016 Nov;166(15-16):487–99. https://doi.org/10.1007/s10354-016-0481-y PMID:27468972
29. Everett AJ, Lowe LA, Wilkinson S. Revision of the structures of (+)-tubocurarine chloride and (+)-chondrocurine. J Chem Soc D. 1970;(16):1020–1. https://doi.org/10.1039/c29700001020.
30. Dagnino J. Historia de los relajantes musculares de síntesis. Relajantes musculares en Anestesia y Terapia Intensiva. Álvarez. Madrid: JA, González F y Bustamante R. Segunda Edición. Arán Ediciones; 2000.
31. Cabarrocas E. Perspectivas futuras de los relajantes musculares. In: Álvarez JA, González F, editors. Relajantes musculares en anestesia y terapia intensiva. Madrid: Editorial Libro del Año; 1996. pp. 561–9.
32. Foldes FF. The mode of action of quaternary ammonium type neuromuscular blocking agents. Br J Anaesth. 1954 Nov;26(6):394–8. https://doi.org/10.1093/bja/26.6.394 PMID:13208908
33. Brown DL, Brock-Utne JG. The first structure-function approach to drug design in anaesthesia. Dr David S. Savage and pancuronium bromide. Anaesthesia. 1997 Dec;52(12):1202–4. https://doi.org/10.1111/j.1365-2044.1997.262-az0401.x PMID:9485976
34. Hewett CL, Savage DS. Amino-steroids. I. 16piperidino-and 16-morpholino-androstanes and -oestra-1,3,5(10)-trienes. J Chem Soc Perkin 1. 1966; 5: 484-8.
35. Hewett CL, Savage DS. Amino-steroids. II. 6-Amino-androstanes and -pregnanes. J Chem Soc Perkin 1. 1967; 7: 582-8.
36. Baird WL, Reid AM. The neuromuscular blocking properties of a new steroid compound, pancuronium bromide. A pilot study in man. Br J Anaesth. 1967 Oct;39(10):775–80. https://doi.org/10.1093/bja/39.10.775 PMID:6073464
37. Boros M, Szenohradszky J, Marosi G, Tóth I. Comparative clinical study of pipecurium bromide and pancuronium bromide. Arzneimittelforschung. 1980;30 2a:389–93. PMID:6248087
38. Savage DS, Sleigh T, Carlyle I. The emergence of NC-45, The emergence of ORG NC-45,1-[(2ß,3a,5a,16ß,17ß)-3,17-bis(acetyloxy)-2-(1-piperi-dinyl)-androstan-16-YL]-1- methylpiperidinium bromide, from the pancuronium series. Br J Anaesth. 1980;52:3S–9S. https://doi.org/10.1093/bja/52.suppl_1.3S PMID:6107102
39. Savarese JJ, Kitz RJ. Does clinical anesthesia need new neuromuscular blocking agents? Anesthesiology. 1975 Mar;42(3):236–9. https://doi.org/10.1097/00000542-197503000-00002 PMID:123132
40. Crul JF, Booij LH. First clinical experiences with ORG NC 45. Br J Anaesth. 1980;52 Suppl 1:49S–52S. https://doi.org/10.1093/bja/52.suppl_1.49S PMID:6107106
41. Agoston S, Salt P, Newton D, Bencini A, Boomsma P, Erdmann W. The neuromuscular blocking action of ORG NC 45, a new pancuronium derivative, in anaesthetized patients. A pilot study. Br J Anaesth. 1980;52 Suppl 1:53S–9S. https://doi.org/10.1093/bja/52.suppl_1.53S PMID:6107107
42. Bowman WC, Rodger IW, Houston J, Marshall RJ, McIndewar I. Structure:action relationships among some desacetoxy analogues of pancuronium and vecuronium in the anesthetized cat. Anesthesiology. 1988 Jul;69(1):57–62. https://doi.org/10.1097/00000542-198807000-00009 PMID:2898902
43. Marshall RJ, Muir AW, Sleigh T, Savage DS. Research and development of aminosteroid neuromuscular blocking agents: past and future. Eur J Anaesthesiol Suppl. 1995 Sep;11 Suppl.11:5–10. PMID:8557007
44. Wierda JM, de Wit AP, Kuizenga K, Agoston S. Clinical observations on the neuromuscular blocking action of Org 9426, a new steroidal non-depolarizing agent. Br J Anaesth. 1990 Apr;64(4):521–3. https://doi.org/10.1093/bja/64.4.521 PMID:2334630
45. Goulden MR, Hunter JM, Editorial I. Rapacuronium (Org 9487): do we have a replacement for succinylcholine? Br J Anaesth. 1999 Apr;82(4):489–92. https://doi.org/10.1093/bja/82.4.489 PMID:10472209
46. Wierda JM, van den Broek L, Proost JH, Verbaan BW, Hennis PJ. Time course of action and endotracheal intubating conditions of Org 9487, a new short-acting steroidal muscle relaxant; a comparison with succinylcholine. Anesth Analg. 1993 Sep;77(3):579–84. https://doi.org/10.1213/00000539-199309000-00026 PMID:8103650
47. Meakin GH, Pronske EH, Lerman J, Orr R, Joffe D, Savaree AM et al. Bronchospasm after rapacuronium in infants and children. Anesthesiology. 2001 May;94(5):926–7. https://doi.org/10.1097/00000542-200105000-00035 PMID:11388548
48. Stenlake JB, Waigh RD, Urwin J, Dewar GH, Coker GG. Atracurium: conception and inception. Br J Anaesth. 1983;55 Suppl 1:3S–10S. PMID:6688014
49. Fisher DM, Canfell PC, Fahey MR, Rosen JI, Rupp SM, Sheiner LB et al. Elimination of atracurium in humans: contribution of Hofmann elimination and ester hydrolysis versus organ-based elimination. Anesthesiology. 1986 Jul;65(1):6–12. https://doi.org/10.1097/00000542-198607000-00002 PMID:3755306
50. Wastila WB, Maehr RB, Turner GL, Hill DA, Savarese JJ. Comparative pharmacology of cisatracurium (51W89), atracurium, and five isomers in cats. Anesthesiology. 1996 Jul;85(1):169–77. https://doi.org/10.1097/00000542-199607000-00023 PMID:8694363
51. Belmont MR, Lien CA, Quessy S, Abou-Donia MM, Abalos A, Eppich L et al. The clinical neuromuscular pharmacology of 51W89 in patients receiving nitrous oxide/opioid/barbiturate anesthesia. Anesthesiology. 1995 May;82(5):1139–45. https://doi.org/10.1097/00000542-199505000-00008 PMID:7741288
52. Savarese JJ, Ali HH, Basta SJ, Embree PB, Scott RP, Sunder N et al. The clinical neuromuscular pharmacology of mivacurium chloride (BW B1090U). A short-acting nondepolarizing ester neuromuscular blocking drug. Anesthesiology. 1988 May;68(5):723–32. https://doi.org/10.1097/00000542-198805000-00010 PMID:2967039

 Creative Commons Attribution
Creative Commons Attribution