Valentina Hernández N. 1 ,2 , Priscilla Ulloa V. 1, 2 , Francisca Cortés A. 1 , Waldo Merino U. 1 ,2
 ORCID
ORCIDRecibido: 12-04-2018
Aceptado: 08-05-2018
©2018 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 47 Núm. 3 pp. 224-228|https://doi.org/10.25237/revchilanestv47n03.09
PDF|ePub|RIS
Respiratory complication in the late postoperative period in a patient with type I myotonic dystrophy. Case report
Abstract
Myotonic dystrophy is an uncommon disease, characterised by disorders of the muscle membrane. Its clinical manifestations are muscle weakness, difficulty at initiating movements and delayed muscle relaxation. Carriers of this disease are very sensitive to anaesthetic drugs. Residual neuromuscular blockade is common among these patients, leaving them at risk of various postoperative complications. Proper neuromuscular blockade reversal is therefore crucial. We report the case of an 18-year-old male with myotonic dystrophy type I (Steinert’s disease), who was admitted for a complicated hydatid cyst. He required a laparotomy, which was done under general anesthesia with no intraoperative incidents. He was extubated at the end of the procedure, with 94% response at the train-of-four (TOF) and adequate spontaneous ventilation. No reversal for neuromuscular blockade was given. The patient evolved favourably during the postoperative phase. However, in the later postoperatory period the patient presented severe respiratory complications. Proper anaesthetic management of these patients, as described in the literature, includes the use of non-depolarising muscle relaxants, monitoring of muscle relaxation and reversal of neuromuscular blockade. The combination of rocuronium and sugammadex appears to convey the optimum reversal required for these cases.
Resumen
Las distrofias miotónicas son enfermedades poco comunes, caracterizadas por trastornos a nivel de la membrana muscular. Clínicamente se manifiestan por debilidad muscular progresiva, dificultad al iniciar movimientos y retardo en la relajación muscular. Los portadores de este grupo de enfermedades tienen una marcada sensibilidad a los fármacos anestésicos. Es habitual que presenten bloqueo neuromuscular residual, arriesgándose a sufrir diversas complicaciones postoperatorias. Por ello, es importante realizar una reversión adecuada de la relajación muscular en estos pacientes. Presentamos el caso de un paciente masculino de 18 años, con distrofia miotónica de Steinert tipo I, que ingresa para laparotomía por quiste hidatídico hepático complicado. Recibió anestesia general sin incidentes. Es extubado con una respuesta al tren-de-cuatro (TOF) de 94% y ventilación espontánea adecuada. No se realiza reversión del bloqueo neuromuscular y evoluciona favorablemente en el postoperatorio inmediato. Sin embargo, en el período postoperatorio tardío, presenta complicaciones respiratorias severas. El adecuado manejo de estos pacientes, según lo recomendado en la literatura, requiere el uso de relajantes no-depolarizantes, monitorización y reversión del bloqueo neuromuscular, siendo probablemente la combinación de rocuronio y sugammadex, la más adecuada para estos fines.
-
Introducción
Las distrofias miotónicas son un grupo de enfermedades infrecuentes, multisistémicas, autosómicas dominantes, caracterizadas por debilidad muscular progresiva, dificultad al iniciar movimientos y retardo en la relajación muscular. Tienen una incidencia de 1/8.000 recién nacidos y una prevalencia de 2,1-14,3/100.000[1],[2]. Aunque pueden manifestarse a cualquier edad, la mayoría de los casos se presenta entre 20-40 años[3],[4],[5],[6],[7].
Estos pacientes son de especial cuidado en el perioperatorio, ya que tienen una notable sensibilidad a los anestésicos. Además, el frío de pabellón puede desencadenar crisis miotónicas, a lo que se asocia complicaciones respiratorias y cardiacas postoperatorias[3],[4],[5].
Es ampliamente conocido que el bloqueo neuromuscular residual aumenta el riesgo de complicaciones respiratorias postoperatorias[5]. En diversas publicaciones, se enfatiza la importancia de realizar una adecuada reversión del bloqueo neuromuscular en pacientes con distrofia miotónica ya que por su afectación muscular y extramuscular tienen mayor riesgo de complicaciones en el postoperatorio[8]. Se debe evitar el uso de relajantes musculares despolarizantes, ya que pueden generar una contracción muscular prolongada sin posibilidad de reversión[7],[9].
A continuación, se presenta el caso de un paciente masculino de 18 años, portador de distrofia miotónica de Steinert tipo I, que ingresa para laparotomía exploratoria por un quiste hidatídico complicado. Evoluciona favorablemente en el postoperatorio inmediato. Sin embargo, presenta complicaciones respiratorias en el postoperatorio tardío.
-
Caso clínico
Hombre de 18 años, 60 kg, 175 cm (índice de masa corporal: 19,59 kg/m2), portador de distrofia miotónica de Steinert tipo I, fumador activo (1 cigarrillo diario), antecedente de hospitalización en Unidad de Tratamiento Intensivo por neumonía adquirida en la comunidad e insuficiencia respiratoria a los 14 años, sin necesidad de ventilación mecánica. Sin otros antecedentes mórbidos, quirúrgicos ni alergias. Actualmente, sin limitación de sus actividades de la vida diaria. Su padre es portador de distrofia muscular con síntomas leves y su hermano mayor presenta síntomas severos.
Ingresa al Servicio de Urgencia del Hospital Hernán Henríquez Aravena con diagnóstico de quiste hidatídico hepático complicado, de 16 cm x 17 cm, con indicación de cirugía urgente. No se identificaron predictores de vía aérea difícil.
En pabellón se realiza monitorización básica con electrocardiograma, saturometría, capnografía, presión arterial no invasiva, temperatura y monitorización de bloqueo neuromuscular (TOF-Watch®S). No se identificaron predictores de vía aérea difícil. Inducción endovenosa con fentanyl 2,5 ug/kg, lidocaína 1 mg/kg, propofol 2,5 mg/kg y rocuronio 0,4 mg/kg. Se realizó intubación endotraqueal sin incidentes mediante laringoscopía directa. Durante la mantención se utilizó O2/N2O 50/50 e isofluorano 1,2%, analgesia con metamizol 1 g y nefersil 100 mg.
Se realizó quistectomía, colecistectomía e instalación de drenaje Jackson Pratt. El tiempo quirúrgico fue 42 min. Fue extubado sin incidentes, con respuesta al TOF de 94% y ventilación espontánea con frecuencia de 16 respiraciones por minuto y CO2 espirado de 30 mmHg. Con estos antecedentes se decide no realizar reversión del bloqueo neuromuscular con
sugammadex. Se mantiene con bomba de infusión continua de analgesia (fentanyl 600 mcg + nefersil 300 mg + metamizol 1 g en 250 ml de suero fisiológico) y se traslada a sala de recuperación.
En el postoperatorio inmediato evolucionó sin incidentes, sin requerimiento de oxígeno, con ventilación y hemodinamia estable. Cumplió criterios de Aldrete a las 2 horas y se trasladó a sala común.
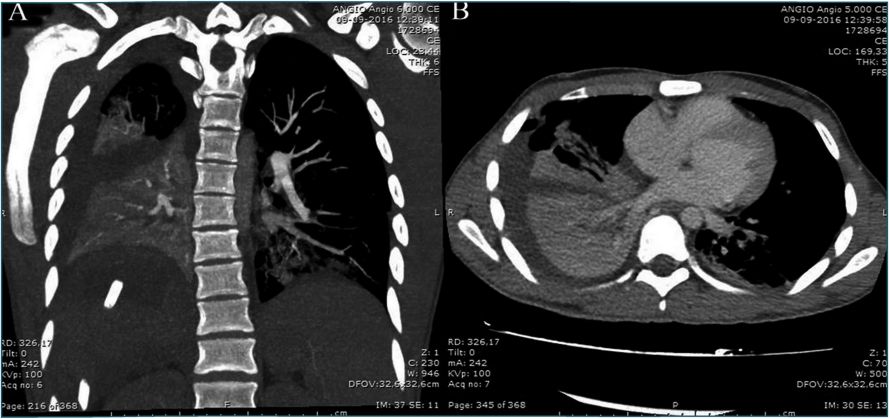
A las 48 horas presentó taquicardia hasta 120 latidos por minuto, disnea, polipnea y desaturación hasta 80%. Al examen físico se observa sudoroso, con retracción de los músculos intercostales, sibilancias bilaterales en dos tiempos, hemitórax derecho con roncus difusos y disminución del murmullo pulmonar en su base. En los exámenes de laboratorio destacó proteína C reactiva: 200 mg/L y leucocitosis: 13,72 K/uL. Se solicitó radiografía y angiografía por tomografía axial computarizada (AngioTAC) que mostraron condensaciones atelectásicas bibasales, de predominio derecho, asociado a derrame pleural ipsilateral y neumoperitoneo (Figura1). Se trasladó a Unidad de Cuidados Intensivos (UCI).
En UCI fue manejado con ventilación mecánica no invasiva, presión inspiratoria pico de 15 y FiO2 0,4 durante 2 días, y kinesioterapia respiratoria intensiva, con lo que se recuperó rápidamente. A los 6 días reingresó a su habitación, persistiendo con requerimiento de oxígeno 3 lts por naricera. Posteriormente, evolucionó en buenas condiciones generales y fue dado de alta a los 10 días desde su ingreso.
 |
|
Figura 1 |
-
Discusión
Las distrofias miotónicas agrupan a enfermedades con trastornos a nivel de la membrana muscular en las que se genera una contracción mantenida después de estimulación neuromuscular. En el caso de la distrofia muscular tipo I de Steinert (clásica), existe predominio de atrofia y debilidad[5],[10],[11].
La distrofia miotónica es una enfermedad crónica, multisistémica, autosómica dominante, causada por una mutación del cromosoma 19 que afecta los genes que codifican una proteína quinasa expresada en el músculo liso, músculo esquelético y miocardio[1],[2] Tienen una mortalidad 7,3 veces superior y menor esperanza de vida que el promedio de la población general[1].
Se ha descrito una mínima interacción del propofol con la función neuromuscular. La recuperación del efecto de estos agentes es rápida, incluso en pacientes con desórdenes neuromusculares[4]. Una infusión controlada a sitio efecto, ha sido considerada como una técnica anestésica adecuada para estos pacientes debido a su conveniente vida media sensitivo contextual y fácil titulación[3].
En cuanto al uso de los gases halogenados, a pesar de tener un mayor potencial de depresión miocárdica, se han utilizado sin inconvenientes[12]. No existe riesgo de hipertermia maligna en esta patología, por lo que no existe contraindicación para su uso[9].
Estos pacientes son muy sensibles a los opioides. Sin embargo, se han utilizado sufentanyl y fentanyl sin incidentes y se han reportado casos de uso de infusión continua de remifentanyl de forma segura. Aun así, en un estudio retrospectivo se describió que un 57% de los pacientes que utilizaron infusión continua de morfina en el postoperatorio desarrollaron complicaciones respiratorias, en comparación con sólo un 6% de complicaciones observadas en aquellos que no la usaron[2].
La distrofia miotónica en su forma clásica, presenta mayor incidencia de complicaciones perioperatorias cardiacas, y respiratorias, como ocurrió en este caso, debido a una aumentada sensibilidad a los agentes no despolarizantes y a una impredecible e inadecuada reversión del bloqueo neuromuscular[4],[5],[10]. Estos pacientes tienen contraindicación de un bloqueo neuromuscular de tipo despolarizante por el riesgo de hiperkalemia, rabdomiólisis y de desencadenar una crisis miotónica y/o paro cardiaco[4],[6]. Existe mayor riesgo de curarización residual postoperatoria y, por tanto, es importante la reversión completa del bloqueo neuromuscular, aún cuando en la monitorización parezca innecesaria como ocurrió en el caso presentado.
Diferentes publicaciones recomiendan el uso de relajantes no despolarizantes como rocuronio, vecuronio y atracurio[7]. En el caso presentado se utilizó rocuronio, el cual alcanza rápidamente un bloqueo neuromuscular apropiado y cuyo uso se ha mostrado seguro en pacientes con desórdenes neuromusculares[4],[12]. El bloqueo neuromuscular inducido por rocuronio se puede revertir con inhibidores de la colinesterasa. Sin embargo, esta forma de reversión es lenta, impredecible, poco confiable y puede llevar a espasmo muscular. El Sugammadex provee una reversión rápida y segura del bloqueo neuromuscular por rocuronio y vecuronio, lo que resulta en una rápida recuperación de la función neuromuscular. La combinación de rocuronio y Sugammadex ha mostrado ser la forma más efectiva de reversión en pacientes con distrofia miotónica. Sus dosis deben ser individualmente optimizadas mediante monitoreo neuromuscular, ya que puede ocurrir recurarización posterior a la administración de una baja dosis de Sugammadex[3].
Es importante recordar las recomendaciones de Murphy and Brull sobre el uso de relajantes musculares, que establecen:[13]
1. Se debe utilizar una monitorización cuantitativa de la transmisión neuromuscular en todo paciente que utilice fármacos bloqueadores neuromusculares.
2. La monitorización de la transmisión neuromuscular con TOF y “Double Bourst Stimulation” no excluye bloqueo neuromuscular residual.
3. Los test clínicos de la función muscular no son fidedignos predictores de la recuperación de la función neuromuscular.
4. Si no se utiliza un monitor neuromuscular o estimulador de nervio periférico, se debe administrar rutinariamente reversión farmacológica y solo cuando la actividad muscular espontánea esté presente.
Las recomendaciones clínicas de la Sociedad de Anestesiología de Chile y otras publicaciones similares sobre monitorización, establecen la necesidad de disponer de un equipo en todo paciente en quien se efectúe bloqueo neuromuscular. Señalan que un TOF-Ratio > 0,9 es el estándar para considerar una apropiada reversión[13],[14],[15],[16].
Hasta el momento, no hemos encontrado casos descritos de pacientes con distrofia miotónica de Steinert en los que se desarrolle una complicación respiratoria en el postoperatorio tardío como en este paciente. Sin embargo, en este caso, es difícil adjudicar esta complicación exclusivamente a la ausencia de reversión neuromuscular en el período quirúrgico. Se debe considerar otros factores que pueden haber influido. Las atelectasias y el derrame pleural descritas en el AngioTAC, pueden explicarse como complicación respiratoria común en cirugía abdominal alta como la quistectomía hepática. Además, se debe considerar que el paciente no fue traslado a una UCI desde la unidad de recuperación, ni recibió kinesioterapia en el período postoperatorio inmediato. Es probable que la complicación descrita sea de origen multifactorial.
-
Referencias
- Catena V, Del Monte DD, Rubini A, Guccione C, Ricagna F, Gangeri G, et al. Anesthesia and myotonic dystrophy (Steinert’s syndrome). The role of total intravenous anesthesia with propofol, cisatracurium and remifentanyl. Case report. Minerva Anestesiol. 2007 Sep;73(9):475–9. PMID:17660741
- Sinclair JL, Reed PW. Risk factors for perioperative adverse events in children with myotonic dystrophy. Paediatr Anaesth. 2009 Aug;19(8):740–7. https://doi.org/10.1111/j.1460-9592.2009.03079.x PMID:19624361
- Kashiwai A, Suzuki T, Ogawa S. Sensitivity to rocuronium-induced neuromuscular block and reversibility with sugammadex in a patient with myotonic dystrophy. Case Rep Anesthesiol. 2012;2012:107952. https://doi.org/10.1155/2012/107952 PMID:22606401
- Stewart PA, Phillips S, De Boer HD. Sugammadex reversal of rocuronium-induced neuromuscular blockade in two types of neuromuscular disorders: myotonic dystrophy and spinal muscular atrophy. Rev Esp Anestesiol Reanim. 2013 Apr;60(4):226–9. https://doi.org/10.1016/j.redar.2012.07.007 PMID:22947194
- Gurunathan U, Duncan G. The successful use of sugammadex and uneventful recovery from general anaesthesia in a patient with myotonic dystrophy. Indian J Anaesth. 2015 May;59(5):325–6. https://doi.org/10.4103/0019-5049.156894 PMID:26019363
- Kirzinger L, Schmidt A, Kornblum C, Schneider-Gold C, Kress W, Schoser B. Side effects of anesthesia in DM2 as compared to DM1: a comparative retrospective study. Eur J Neurol. 2010 Jun;17(6):842–5. https://doi.org/10.1111/j.1468-1331.2009.02942.x PMID:20100232
- Owen PM, Chu C. Emergency caesarean section in a patient with myotonic dystrophy: a case of failed postoperative extubation in a patient with mild disease. Anaesth Intensive Care. 2011 Mar;39(2):293–8. PMID:21485681
- Subramaniam A, Grauer R, Beilby D, Tiruvoipati R. Anesthetic management of a myotonic dystrophy patient with paraganglionoma. J Clin Anesth. 2016 Nov;34:21–8. https://doi.org/10.1016/j.jclinane.2016.03.035 PMID:27687340
- White RJ, Bass SP. Myotonic dystrophy and paediatric anaesthesia. Paediatr Anaesth. 2003 Feb;13(2):94-102. Review. PubMed PMID: 12562480. https://doi.org/10.1046/j.1460-9592.2003.00889.x.
- Weingarten TN, Hofer RE, Milone M, Sprung J. Anesthesia and myotonic dystrophy type 2: a case series. Canadian Journal of Anesthesia/Journal canadien d’anesthésie [Internet]. Springer Nature; 2010 Jan 15;57(3):248–55. Available from: http://dx.doi.org/10.1007/s12630-009-9244-1
- Álvarez, JA, González F, Bustamante R, Relajantes musculares en anestesia y terapia intensiva, 2da ed, España, Arán, 2000, pp.608.
- Veyckemans F, Scholtes JL. Myotonic dystrophies type 1 and 2: anesthetic care. Paediatr Anaesth. 2013 Sep;23(9):794–803. https://doi.org/10.1111/pan.12120 PMID:23384336
- Brull SJ, Murphy GS. Residual neuromuscular block: lessons unlearned. Part II: methods to reduce the risk of residual weakness. Anesth Analg. 2010 Jul;111(1):129–40. https://doi.org/10.1213/ANE.0b013e3181da8312 PMID:20442261
- Bulka CM, Terekhov MA, Martin BJ, Dmochowski RR, Hayes RM, Ehrenfeld JM. Nondepolarizing Neuromuscular Blocking Agents, Reversal, and Risk of Postoperative Pneumonia. Anesthesiology. 2016 Oct;125(4):647–55. https://doi.org/10.1097/ALN.0000000000001279 PMID:27496656
- Fortier LP, McKeen D, Turner K, de Médicis É, Warriner B, Jones PM, et al. The RECITE Study: A Canadian Prospective, Multicenter Study of the Incidence and Severity of Residual Neuromuscular Blockade. Anesth Analg. 2015 Aug;121(2):366–72. https://doi.org/10.1213/ANE.0000000000000757 PMID:25902322
- Murphy GS, Brull SJ. Residual neuromuscular block: lessons unlearned. Part I: definitions, incidence, and adverse physiologic effects of residual neuromuscular block. Anesth Analg. 2010 Jul;111(1):120–8. https://doi.org/10.1213/ANE.0b013e3181da832d PMID:20442260