Gonzalo Ferrer Moreno1, Rebecca Fernández Arredondo2*
 ORCID
ORCIDRecibido: 16-01-2024
Aceptado: 02-09-2024
©2025 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 54 Núm. 2 pp. 217-220|https://doi.org/10.25237/revchilanestv54n2-17
PDF|ePub|RIS
Abstract
Pierre Robin Sequence (PRS) is a congenital condition that affects infants, characterized by micrognathia, glossoptosis, and airway obstruction due to an abnormal development of the first branchial arch. Although it used to be associated with cleft palate, this feature is no longer mandatory for the diagnosis since 2014. With autosomal recessive inheritance, PRS has an incidence of 1:3,000 to 1:30,000 live births and is often associated with other congenital syndromes. Patients address systemic challenges, such as cardiac and neurological problems. Airway obstruction and feeding difficulties are characteristic issues. Management involves both non-surgical approaches (prone positioning, CPAP) and surgical interventions (tongue adhesion, osseous mandibular distraction). The complexity of PRS emphasize the need for algorithms for a safe and effective approach, improving the quality of life for patients.
Resumen
La Secuencia de Pierre Robin (SPR) es una condición congènita que afecta a lactantes, caracterizada por micrognatia, glosoptosis y obstrucción de las vías respiratorias debido al desarrollo anormal del primer arco branquial. Aunque históricamente asociada con paladar hendido, esta característica ya no es obligatoria para el diagnóstico desde 2014. Con herencia autosómica recesiva, la SPR tiene una incidencia de 1:3.000 a 1:30.000 nacimientos vivos y a menudo se asocia con otros síndromes congénitos. Los pacientes enfrentan desafíos sistémicos, como problemas cardíacos y neurológicos. La obstrucción de las vías respiratorias y las dificultades en la alimentación son problemas destacados. El manejo implica enfoques no quirúrgicos (posición prona, CPAP) y quirúrgicos (adhesión de la lengua, distracción mandibular ósea). La complejidad de la SPR subraya la necesidad de algoritmos para un enfoque seguro y efectivo, mejorando la calidad de vida de los pacientes.
-
Caso clínico
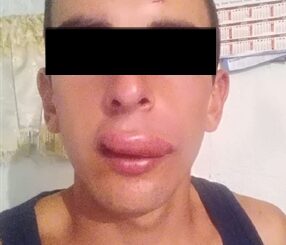
Mediante interrogatorio indirecto (madre) se obtiene la siguiente información: masculino, 5 años 4 meses 2 días de edad (fecha de nacimiento: 16 de marzo de 2018). Antecedentes heredofamiliares: rama materna con carga genética para diabetes mellitus tipo 2 y sobrepeso, rama paterna con antecedentes interrogados y negados; Antecedentes personales no patológicos: hemotipo O+, inmunizaciones incompletas, se corroboran datos mostrando la cartilla nacional de vacunación (México), exposición a biomasa o tabaquismo pasivo interrogado y negado, dieta integrada a la familiar sin restricciones alimenticias, refiere dificultad para la succión, niega datos sugerentes de apnea obstructiva del sueño, niega datos de epilepsia, niega asma o hiperreactividad bronquial. Antecedentes perinatales: producto de tercera gestación nacido por cesárea iterativa, se obtuvo producto único vivo, con APGAR 8 y Silverman 0 al minuto de nacido, y APGAR 9 y Silverman de 0 a los 5 minutos de nacido, a la revisión física se encuentra dismorfia facial (micrognatia y paladar hendido) y signos de dificultad respiratoria (tiraje intercostal, disociación toracoabdominal y cianosis peribucal), es asistido con oxígeno a cinco litros por minuto mediante casco cefálico, se le coloca sonda orogástrica y se mantiene hospitalizado en observación durante dos días, al presentar aparente mejoría, se retira sonda orogástrica y egresa a domicilio con indicación de alimentación exclusiva mediante biberón para paladar hendido con fórmula maternizada. A las 24 h la madre refiere la presencia de palidez mucotegumentaria y signos de dificultad respiratoria a la alimentación (silbído espiratorio, tiraje intercostal, cianosis peribucal) que remiten con el cambio de posición (a posición prona) por lo que acude al servicio de urgencias en Tapachula, Chiapas, México donde es ingresado a hospitalización para optimizar la oxigenación mediante el uso de casco cefálico; en el séptimo día de hospitalización previo a la instauración de una segunda sonda orogástrica el paciente presentó siete eventos de paro cardiorrespiratorio para un total de tiempo acumulado de veinte minutos, se le comentó a la madre el fracaso de abordaje de la vía aérea. Se traslada el paciente al hospital de ISSSTE Zaragoza en Ciudad de México, en donde lo recibieron: hipoactivo, con signos de dificultad respiratoria y con apoyo de oxígeno mediante casco cefálico. Se realizó interconsulta con el servicio de anestesiología y mediante una laringoscopía diagnóstica se orotintuba con técnica mixta mediante fibros- copio en apoyo con videolaringoscopio con tubo endotraqueal tipo murphy sin balón de calibre 3.0, sin incidentes. Posterior al aseguramiento de vía aérea se decide realizar: tomografía com- putarizada de cráneo, electroencefalograma, radiografía de tórax, electrocardiograma de doce derivaciones en los cuales se reporta: leucomalasia, datos sugerentes de neumonía química, atelectasias basales unilaterales derechas difusas. Ingresó a la unidad de terapia intensiva permaneciendo orointubado durante 2 meses, agregándose: anemia ferropénica, enterocolitis necrotizante, sangrado de tubo digestivo bajo los cuales son manejados con apoyo aminérgico durante quince días a base de dobutamina y adrenalina dosis respuesta; sulfato ferroso, eritropoyetina, transfusión de concentrados eritrocitarios, una dosis de surfactante pulmonar intramuscular, fototerapia, múltiples esquemas de antibióticoterapia y la colocación de una gastrostomía percutanea modificada con sonda Pezzer. Se progresó de ventilador a modo CPAP hasta tolerar ventilación espontánea, se decide extubación y apoyo con oxígeno mediante puntas nasales, al ser exitosa y tolerable la vía oral, se le retiró gastrostomía. Egresó del hospital a los dos meses y cincuenta y dos días de nacido, acudiendo a seguimiento durante el primer año de vida en su clínica de adscripción en donde se realizó diagnóstico de SPR y retraso en el desarrollo neurológico secundario a hipoxia, retraso en el desarrollo del lenguaje debido a la fisura palatina. Se pierde seguimiento hasta los cuatro años de vida, momento en el cual es referido al Centro Médico Nacional 20 de Noviembre en Ciudad de México, para la corrección de fisura incompleta de paladar. Ingresa a cargo del servicio de cirugía plástica. En la exploración física se encuentra paciente de 5 años de edad con peso de 22 kg y 121 cm de altura, temperatura de 36,2 °C, frecuencia respiratoria de 22 respiraciones por minuto, frecuencia cardíaca de 109 latidos por minuto, presión arterial de 100/52 mmHg, saturación de oxígeno del 92% al aire ambiente. Paciente activo, reactivo, poco cooperador con datos de ansiedad por separación, glasgow modificado de 15 puntos con adecuada coloración de tegumentos y adecuada hi- dratación de mucosas, cráneo braquicefálico con adecuada implantación de pabellones auriculares y línea de cabello, pupilas isocóricas normorefléxicas, pabellón nasal amplio, con narinas permeables mandíbula con micrognatia, cavidad oral sin limitación para la apertura, piezas dentales incompletas, presencia de primeros y segundos molares bilaterales superiores e inferiores en mal estado, caninos centrales y laterales inferiores presentes, vestíbulo con la presencia de fisura que compromete paladar duro y se extiende más allá de orofaringe, úvula aparentemente bífida, hipertrofia amigdalina grado I, incapacidad de fonación adecuada, cardiopulmonar sin compromiso evidente, abdomen blando, plano, depresible, normoperistalsis, sin datos de abdomen agudo, extremidades íntegras, simétricas, con adecuados arcos de movimiento, pulsos presentes, llenado capilar de 2 segundos. Se recaban exámenes de laboratorio del día 25 de julio de 2023: química sanguínea: BUN 13 mg/dl, glucosa 82 mg/dl, creatinina sérica 0,45 mg/dl, relación BUN/creatinina 28,9. electrolítos séricos: sodio 137 mEq/L, potasio 4,3 mEq/L, bilirrubina total 0,31 mg/dl, bilirrubina indirecta 0,21 mg/dl, bilirrubina directa 0,10 mg/dl. Tiempo de tromboplastina parcial 21,2 seg, tiempos de trombina 20,4 seg, tiempo de protrombina 10,6 seg, INR 0,9. Biometría hemática: leucocitos 6,54 miles/mm3, hemoglobina 13,1 g/dl, hematocrito 40,0%, plaquetas 314 mi- les/mm3. Se otorga riesgo anestésico ASA E III A, se programa para palatoplastía bajo anestesia general balanceada. Se explica a la madre la técnica anestésica sugerida, posibles riesgos y complicaciones asociadas, se resuelven dudas y firma consentimiento informado, se solicitan ocho horas de ayuno para sólidos y dos horas para líquidos claros. Se planea técnica de intubación mixta con fibroscopio y videolaringoscopio.
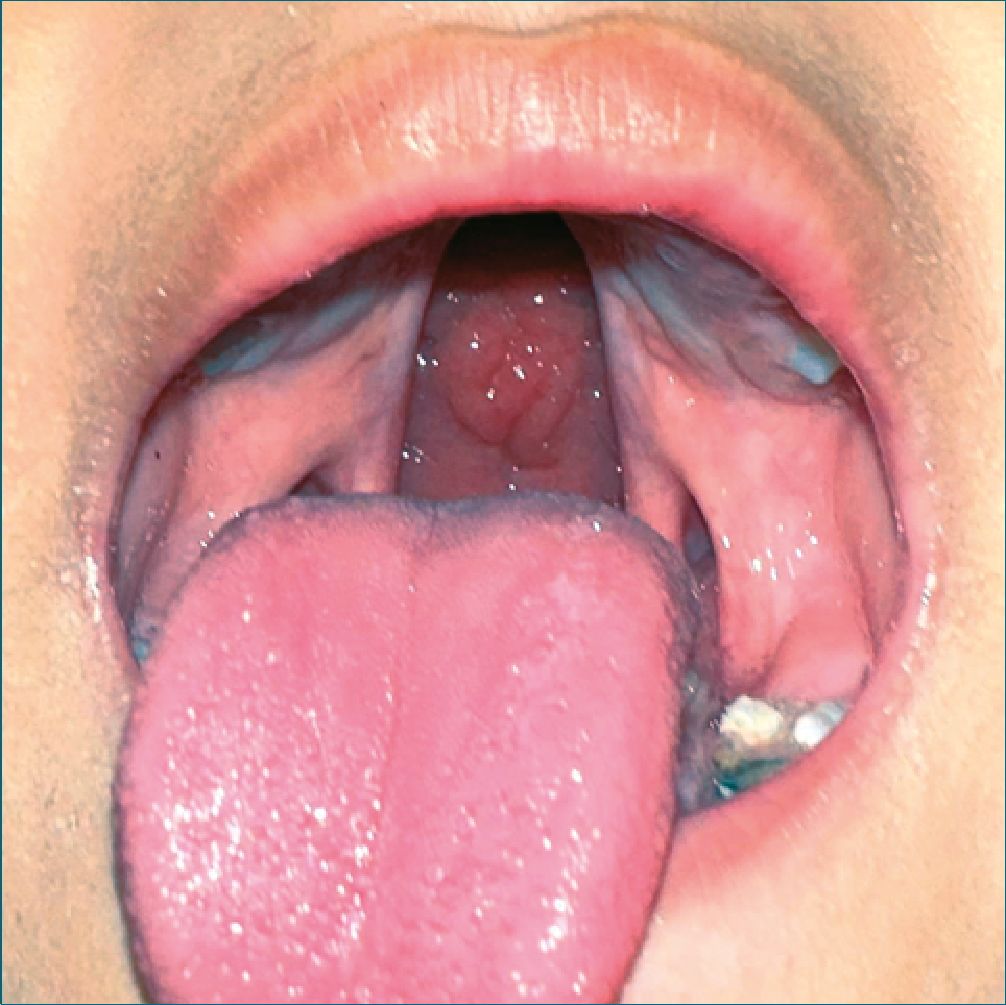
Figura 1.
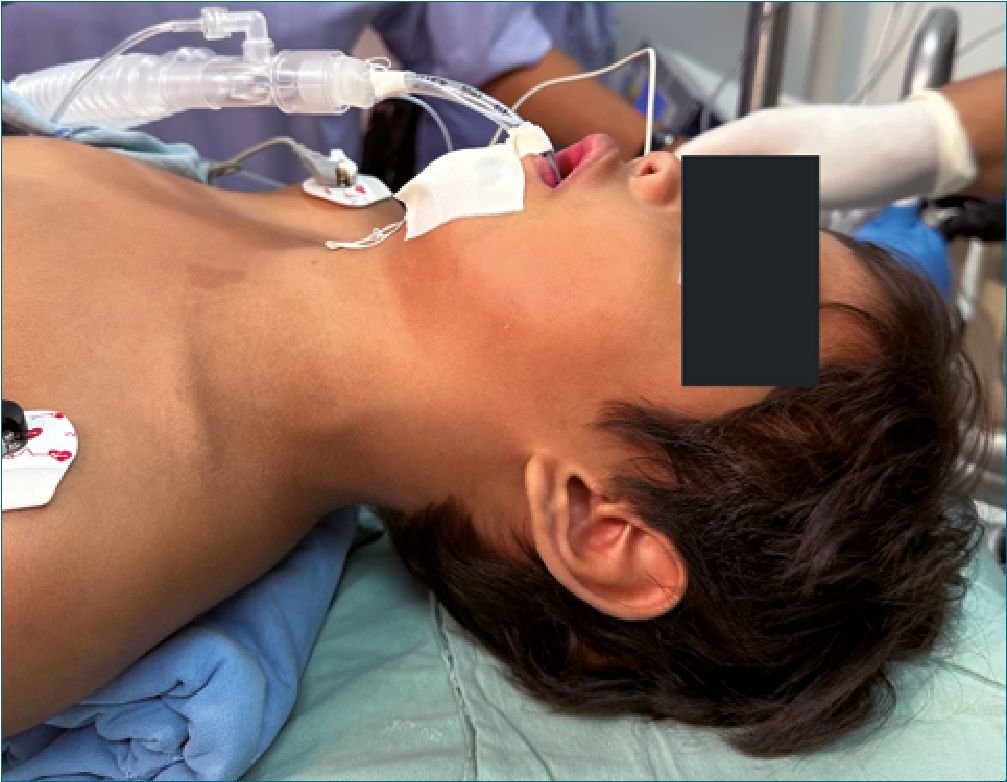
Figura 2.
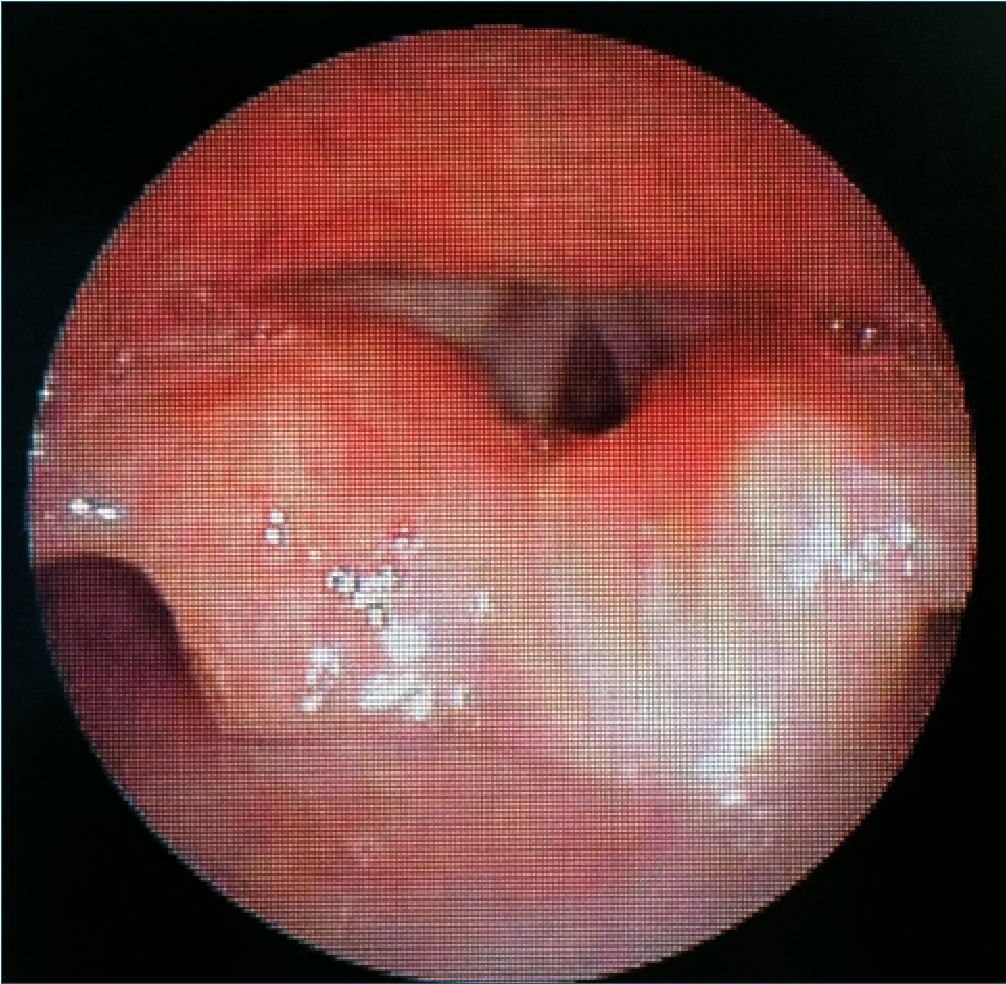
Figura 3.
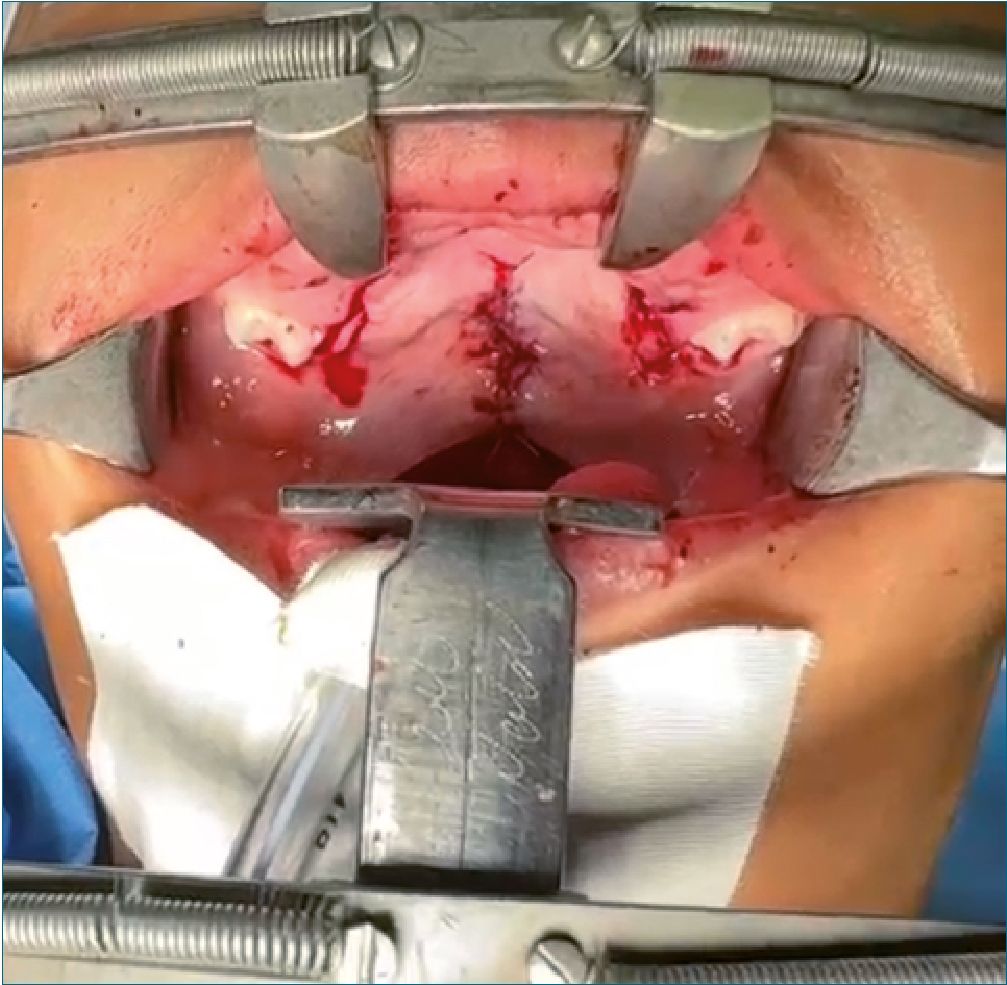
Figura 4.
-
Técnica anestésica
Se prepara paciente para quirófano el día 18 de agosto de 2023, se recibe paciente consciente, poco cooperador, con adecuada hidratación de mucosas y coloración de tegumentos, se corrobora máquina de anestesia y equipo de vía aérea disponible, se realiza monitorización no invasiva y colocación de BIS: presión arterial: 101/61 mmHg, frecuencia cardíaca: 92 latidos por minuto, frecuencia respiratoria: 19 respiraciones por minuto, saturación de oxígeno: 92% al aire ambiente, BIS de 95, se premedica paciente con 2 mg intravenosos (IV) de midazolam, se preoxigena con mezcla de aire/O2 a 5 lt/min para una FiO2 del 50%, se administran 50 mcg IV de fentanil, se otorga latencia de 7 minutos, se administran 30 mg IV de propofol; el paciente se mantiene con ventilación espontánea y oxigenación apnéica a 5 lt/min y FiO2% del 100%, se realiza videolaringoscopía con hoja MAC 2, sin embargo, debido a la asimetría bucal no es posible la introducción efectiva de la hoja del videolaringoscopio, por lo que se ventila a través de mascarilla facial, se administran 20 mg IV de propofol, sin perder el automatismo ventilatorio, se decide la introducción del fibrobroncoscopio con un tubo endotraqueal tipo murphy 4,5 hacia la hipofaringe, al visualizar cuerdas vocales se administran 30 mg de lidocaína 1% simple sobre vía aérea, se desliza tubo endotraqueal y se complementa inducción con 30 mg IV de propofol, 25 mcg IV de fentanil y 4 mg IV de cisatracurio. Se corrobora adecuada colocación de tubo endotraqueal mediante la expansión simétrica de ambos hemitórax, auscultación pulmonar simétrica, presencia de columna de aire en tubo endotraqueal, movimientos de ample- xión y amplexación bilaterales, se conecta tubo endotraqueal a ventilador controlado por presión y se fija tubo endotraqueal a 16 cm. El mantenimiento anestésico se logró con sevoflurane a 2 vol%, infusión endovenosa de fentanil a 0,039 – 0,065 mcg/ kg/min para una concentración plasmática final de 3.6 ng/ml, adyuvantes intravenosos: 330 mg de paracetamol, 5,5 mg de dexametasona, 3,3 mg de ondansetrón, 15 mg de ketorolaco. El procedimiento quirúrgico tuvo como duración una hora y treintaicinco minutos, sin incidentes anestésicos. Una vez finalizado el procedimiento quirúrgico se aspiran secreciones y se progresa paciente a ventilación espontánea, una cumplidos los criterios para extubación, se retira tubo endotraqueal en inspiración profunda sin incidentes, se asiste con mascarilla facial y mezcla de aire a 5 lt/min y FiO2 al 80%. Se traslada paciente a unidad de cuidados postanestésicos con los siguientes signos vitales: presión arterial 92/48 mmHg, frecuencia cardíaca 92 latidos por minutos, frecuencia respiratoria de 18 respiraciones por minuto y saturación de oxígeno de 93% al aire ambiente, Aldrete 9/10, FLACC 1/10, y Stewart 5/6
-
Discusión
La Secuencia de Pierre Robin (SPR) es una condición con- génita rara que afecta a los lactantes y se caracteriza por la presencia de micrognatia, glosoptosis y obstrucción de las vías respiratorias[1]. Este trastorno fue inicialmente descrito por el médico holandés Von Siebold en 1835 y posteriormente detallado por el dentista francés Pierre Robin en 1923[1]. Aunque históricamente se asoció con paladar hendido, en 2014, se llegó a un consenso en los Países Bajos que excluyó esta característica como obligatoria para el diagnóstico[2].
La SPR se origina por el desarrollo anormal de las estructuras del primer arco branquial y tiene una herencia autosómica recesiva[3]. Aunque es una condición rara, su incidencia varía de 1:3.000 a 1:30.000 nacimientos vivos[2],[4]. Puede presentarse de manera aislada o asociada con otros síndromes congénitos, siendo el Síndrome de Stickler el más común[3],[5].
Los lactantes con SPR enfrentan desafíos significativos, principalmente relacionados con la obstrucción de las vías respiratorias y las dificultades en la alimentación[6]. El manejo de la SPR se centra en enfoques no quirúrgicos, como la posición prona, el uso de sonda nasofaríngea y presión positiva continua de las vías respiratorias (CPAP)[7],[8]. Además, se consideran abordajes quirúrgicos como la glosopexia, distracción mandibular ósea o, en casos extremos, la traqueostomía[5],[9].
Dada la complejidad de la SPR, es crucial un enfoque multidisciplinario que aborde no solo los problemas respiratorios y nutricionales, sino también otras posibles anomalías sistémicas asociadas[10]. El manejo efectivo de esta condición implica una evaluación exhaustiva y el desarrollo de algoritmos que guíen un enfoque seguro y eficaz para abordar la vía aérea, optimizando la eficiencia y la comunicación en el manejo de estas complejas anomalías congénitas
-
Conclusión
El tratamiento de la SPR requiere una evaluación individualizada debido a la variabilidad en la obstrucción de la vía aérea. Enfoques quirúrgicos, que conllevan anestesia general, presentan riesgos, especialmente en pacientes con SPR. El manejo multidisciplinario basado en protocolos de vía aérea difícil y la experiencia en fibrobroncoscopía son cruciales. En un caso particular, la planificación anticipada permitió reconocer la imposibilidad de utilizar el videolaringoscopio debido a la asimetría facial y fisura palatina. La introducción directa del fibroscopio, planeada por el equipo quirúrgico, facilitó la intubación exitosa sin complicaciones. Se destaca la importancia de una evaluación exhaustiva de la vía aérea antes de procedimientos avanzados y la necesidad de garantizar una ventilación adecuada antes de la extubación en casos de SPR para evitar complicaciones.
-
Referencias
1. Hsieh ST, Woo AS. Pierre Robin Sequence. Clin Plast Surg. 2019 Apr;46(2):249–59. https://doi.org/10.1016/j.cps.2018.11.010 PMID:30851756
2. Zaballa K, Singh J, Waters K. The management of upper airway obstruction in Pierre Robin Sequence. Paediatr Respir Rev. 2023 Mar;45:11–5. https://doi.org/10.1016/j.prrv.2022.07.001 PMID:35987882
3. Jakobsen LP, Ullmann R, Christensen SB, Jensen KE, Mølsted K, Henriksen KF, et al. Pierre Robin sequence may be caused by dysregulation of SOX9 and KCNJ2. J Med Genet. 2007 Jun;44(6):381–6. https://doi.org/10.1136/jmg.2006.046177 PMID:17551083
4. Bush PG, Williams AJ. Incidence of the Robin anomalad (Pierre Robin syndrome). Br J Plast Surg. 1983 Oct;36(4):434–7. https://doi.org/10.1016/0007-1226(83)90123-6 PMID:6626822
5. Gomez-Ospina N, Bernstein JA. Clinical, cytogenetic, and molecular outcomes in a series of 66 patients with Pierre Robin sequence and literature review: 22q11.2 deletion is less common than other chromosomal anomalies. Am J Med Genet A. 2016 Apr;170A(4):870–80. https://doi.org/10.1002/ajmg.a.37538 PMID:26756138
6. Navarrete, S. V. M., Galarza, M. V. V., & Pinto, X. R. M. (2021). Manejo de vía aérea difícil en secuencia de Pierre Robin. Reporte de un caso. Metro Ciencia (En línea). https://doi.org/10.47464/MetroCiencia/vol29/1/2021/44-50.
7. Singer L, Sidoti EJ. Pediatric management of Robin sequence. Cleft Palate Craniofac J. 1992 May;29(3):220–3. https://doi.org/10.1597/1545-1569_1992_029_0220_pmors_2.3.co_2 PMID:1591254
8. Isaza A, Cristancho LA. Oxigenación apneica en el paciente crítico: desde la fisiología hasta la controversia. Acta Colombiana de Cuidado Intensivo; 2020. https://doi.org/10.1016/j.acci.2019.09.003.
9. Johnson GM, Todd DW. Antes 7 Johnson G.M., Todd D.W.: cor pulmonale in severe Pierre Robin syndrome. Pediatrics. 1980;65(1):152–4. https://doi.org/10.1542/peds.65.1.152.
10. Shprintzen RJ. Palatal and pharyngeal anomalies in craniofacial syndromes. Birth Defects Orig Artic Ser. 1982;18(1):53–78. PMID:7115914