Raúl Pizarro R. MD.1,2 Sebastián Vargas2, Diego Varas R. MD.1,2
 ORCID
ORCIDRecibido: 16-12-2024
Aceptado: 26-06-2025
©2026 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 55 Núm. 1 pp. 3-9|https://doi.org/10.25237/revchilanestv55n1-01
PDF|ePub|RIS
Arrhythmogenic right ventricular dysplasia, perioperative management
Abstract
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) is an infrequent hereditary disorder characterized by the replacement of parts of the right ventricular myocardium with fibrofatty tissue, predisposing individuals to ventricular arrhythmias and sudden cardiac death. While well-documented in certain populations, ARVD/C remains under-recognized in Latin America, presenting challenges in perioperative management. The objective of this review is present to the readers key aspects of the etiology, clinical progression, and perioperative management of ARVD/C, with an emphasis on anesthetic implications.
Resumen
La displasia/cardiomiopatía arritmogénica del ventrículo derecho (DAVD/C) es un trastorno hereditario poco frecuente caracterizado por el reemplazo del miocardio del ventrículo derecho por tejido fibro-graso, predisponiendo a arritmias ventriculares y muerte súbita. Aunque bien documentada en ciertas poblaciones, la DAVD/C es poco reconocida en América Latina, presentando desafíos en el manejo perioperatorio. El objetivo de esta revisión es presentar aspectos de la etiología, progresión clínica y manejo perioperatorio de la DAVD/C, con énfasis en implicaciones anestésicas.
-
Introducción
La displasia arritmogénica del ventrículo derecho (DAVD) o cardiomiopatía arritmogénica del ventrículo derecho (CAVD), dentro de las cardiomiopatías hereditarias, es la principal causa de arritmias y una de las causas más frecuentes de muerte súbita en jóvenes. Esta enfermedad se caracteriza por el reemplazo del tejido miocárdico por material fibro-graso, predominando en el ventrículo derecho (VD). Esta patología
predispone a los pacientes a arritmias ventriculares graves y a una disfunción ventricular progresiva, lenta e irreversible[1].
La prevalencia de la DAVD varía según la muestra en estudio. En la población caucásica la prevalencia fluctúa entre 1:1.000 y 1:5.000[1],[2]. En algunas regiones de Italia (Veneto) y Grecia (Isla de Naxos) se ha reportado hasta 8% de prevalencia. En la población latinoamericana la DAVD continúa siendo una patología poco frecuente, aunque su prevalencia exacta no se ha precisado. La enfermedad tiende a manifestarse clínicamente entre la tercera y quinta década de vida, afectando a los hombres tres veces más que a las mujeres[1]-[3].
El objetivo de esta revisión es ofrecer una visión integral sobre las características claves de la fisiopatología de la enfermedad y hacer un enfoque hacia el manejo perioperatorio de los pacientes con displasia arritmogénica de ventrículo derecho (DAVD/C), destacando los desafíos únicos que esta patología presenta, proporcionando información útil y actualizada para la práctica clínica.
-
Material y Métodos
Se realizó una búsqueda exhaustiva en las bases de datos PUBMED, SCOPUS y SPRINGER utilizando las palabras clave: “Arrhythmogenic Right Ventricular Dysplasia AND Anesthesia”, “Arrhythmogenic Right Ventricular Dysplasia AND Perioperative Period”[Mesh] OR Perioperative Care. La búsqueda se limitó a artículos publicados hasta abril de 2025 en idiomas inglés, español, francés y portugués, se logró encontrar 14 artículos el más antiguo publicado en el año 2000 y el más reciente respecto al tema publicado año 2020.
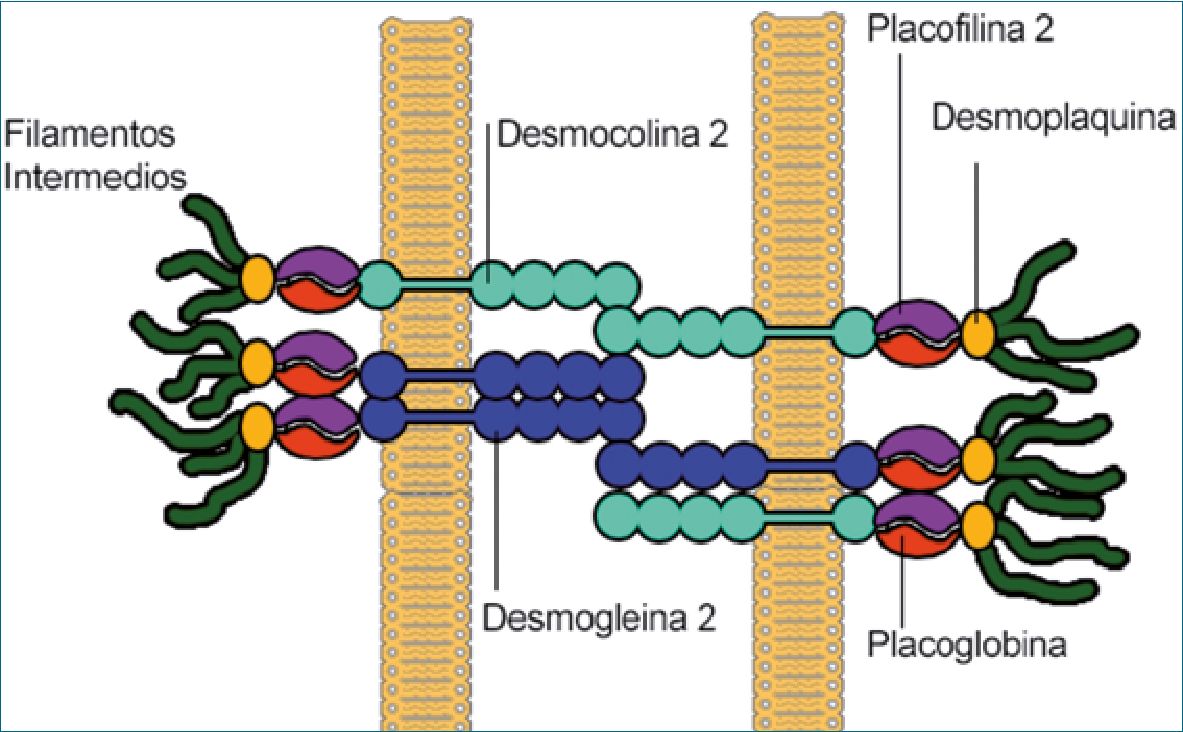
Figura 1. Displasia arritmogenica.
-
Etiología
La DAVD es una cardiomiopatía hereditaria que presenta un patrón hereditario en aproximadamente el 50% de los casos, caracterizándose por una herencia autosómica dominante de penetrancia variable[4]. Sin embargo, existen formas genéticamente recesivas, como el “Síndrome de Naxos” y el “Síndrome de Carvajal”[5]. En ambas presentaciones, los estudios genéticos han identificado mutaciones con una prevalencia 30%-40% placofilina-2 (PKP2),10%-15% desmoplaquina (DSP), 3%-8% desmogleína-2 (DSG2), 1%-5% desmocolina-2 (DSC2) y < 1% placoglobina (JUP), presentando alteraciones en brazos de cromosomas 12, 6, 18 y 17[4]-[6]. Encargadas de codificar proteínas estructurales de los desmosomas miocárdicos (Figura 1). Un estudio de Roudijk et al. (2022)[7], en una cohorte de pacientes con DAVD encontró mutaciones de PKP2 y DSG2 en el 82% de los casos. Estas mutaciones alteran la síntesis
de “dichas proteínas”, lo que afecta la adhesión entre los cardiomiocitos, alterando la estabilidad e integridad del tejido, promoviendo su separación y posterior muerte celular, dando lugar a cicatrización y reemplazo por tejido fibro-graso[8]. Este proceso crea zonas de enlentecimiento y alteración de la conducción eléctrica, lo que favorece la aparición de sitios de reentrada, generando sustrato para la generación de arritmias[9].
-
Historia natural de la enfermedad
Siendo diagnosticado entre la 3ra y 5ta década de la vida, el debut en edades más tempranas o tardías es raro. En el subgrupo con patrón de herencia dominante se ha logrado describir 4 etapas según alteraciones histológicas y sintomatología de los pacientes[10].
Fase Oculta: Subclínica, donde el paciente se encuentra asintomático, con cambios estructurales mínimos o sin ellos, en esta fase la muerte súbita puede ser la primera manifestación.
Fase Arritmica: Presencia de palpitaciones, sincopes, arritmias ventriculares sintomáticas originadas en VC, que son gatilladas al esfuerzo. Es posible encontrar un espectro desde ectopias ventriculares no sostenidas, hasta la muerte súbita por fibrilación ventricular.
Falla de ventrículo derecho: Progresión de remplazo de tejido fibro-graso en VD, llevando a un deterioro de la función sistólica/diastólica del VD, hasta la progresión en una insuficiencia cardíaca derecha.
Falla bi-ventricular: Etapa avanzada de modificaciones estructurales, viéndose afectado por esta infiltración el septum interventricular. También, puede ocurrir trombosis mural, VD aneurismático. En esta etapa se asemeja bastante a una cardiomiopatía avanzada, por lo que en pacientes que llegan a esta etapa el diagnóstico de DAVD/C se ve oculto con otras entidades clínicas.
Se describen 3 zonas clásicas de infiltración durante la progresión de la enfermedad, pared libre de VD a nivel del tracto de entrada VD (sub-tricuspidea), tracto de salida de VD, y ápex recibiendo el nombre de “Triángulo de la displasia”[8].
En patrones de herencia recesiva como en el síndrome de Naxos o Carvajal, no siempre se ve esta progresión, ocurriendo infiltración en anillo mitral, o ventricular dentro de las primeras manifestaciones[5],[8],[11].
-
Diagnóstico
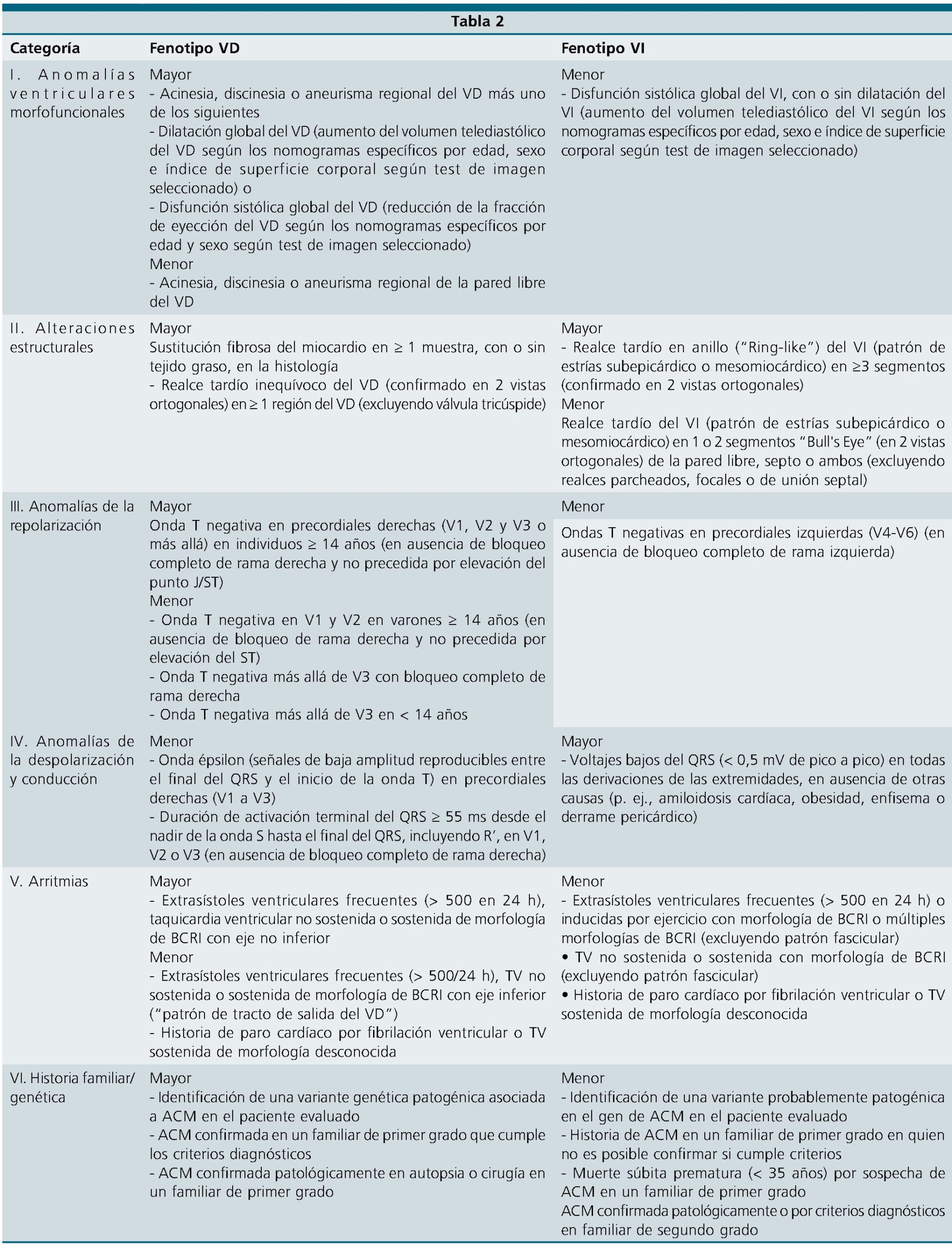
Diagnóstico definitivo es realizado en base a hallazgos histológicos en una biopsia miocárdico, pero dada a la dificultad y morbilidad de realizar esta técnica históricamente se ha realizado el diagnóstico en base a hallazgos clínicos, electrocardiográficos, imagenológicos, estudio genético. Actualmente, existen 2 publicaciones para guiar el diagnóstico, Task Force para el diagnóstico de DAVD/C estadounidense del 2010[12] y Task Force europea para el diagnóstico DAVD/C de 2023, en ambas el diagnóstico es realizado en base a cumplir un número de criterios mayores o menores en diferentes en apartados previamente mencionados. Clasificando el diagnóstico en posible, limite/borderline, definitivo (Tabla 1)[13].
-
Tratamiento
El tratamiento de la DAVD se basa en tres pilares fundamentales: disminuir la mortalidad, ralentizar la progresión de la enfermedad y abolir los síntomas.
La decisión terapéutica se toma evaluando el riesgo de inestabilidad eléctrica ventricular grave, según Corrado et al., describe una estratificación de riesgo en 3 categorías, riesgo bajo, intermedio y alto, de presentar riesgo de muerte o taquiarritmias ventriculares, con el fin de detectar de manera precoz pacientes que se beneficiarían de instalación de un desfibrilador cardioversor implantable (DCI)[14].
Según lo señalado en esta categorización se debe iniciar diferentes medidas.
Cambios de estilos de vida
La restricción de actividad deportiva es el principal factor a considerar, restringiendo cualquier deporte competitivo o de resistencia, permitiendo solamente actividad de baja intensidad. En personas clasificadas como bajo riesgo esta medida puede el único tratamiento para disminuir la progresión de enfermedad[14].
Tratamiento farmacológico
Indicado para manejo de pacientes clasificados con riesgo intermedio a alto, esta dado principalmente por el uso de drogas antiarrítmicas como la amiodarona sola o en conjunto a beta-bloqueo en la mayoría de los pacientes dado a su efecto sinérgico. El uso de otros medicamentos como inibidores de la enzima convertidora de angiotensina (iECA) antagonistas de receptor de angiotensina II (ARA-II), inhibidores de canales de calcio y nitrato no están indicados de regla pero pueden ser útil en quienes presentan daño estructural y/o síntomas de insuficiencia cardíaca[14].
Ablación eléctrica/cateterismo cardíaco
Indicado en pacientes de alto riesgo con TV continuas, o portadores de DCI que presentan shocks de manera adecuada pese al uso de drogas antiarritmicas. Esta terapia se vuelve pilar complementario en estos pacientes dado a que con la progresión del cuadro ocurre la aparición de diferentes focos arritmogenicos[14].
Desfibrilador cardioversor implantable
Terapia más aceptada en pacientes de riesgo alto, generalmente de tipo unicameral, no existen trabajos prospectivos que muestren aumento de la sobrevida dado a esta terapia, sin embargo, la disminución de eventos arritmogenicos potencialmente fatales es indiscutible[14].
Trasplante de corazón
Escenario raro, pero indicado en aquellos pacientes que presentan insuficiencia cardíaca grave sin respuesta a tratamiento médico, de resincronización o en pacientes con arritmias intratables (TV incesantes o tormenta de FV refractaria a terapia de ablación/resincronización)[14].
-
Manejo perioperatorio
Actualmente, no existen recomendaciones ni guías clínicas específicas para el manejo perioperatorio de los pacientes con DAVD, por lo que la mayoría de la literatura disponible se extrapola de otras cardiomiopatías. Dado que la DAVD ha sido reconocida como una causa de muerte súbita no anticipada en el posoperatorio[15], y debido a su progresión con el tiempo, es crucial realizar un abordaje completo en la fase preoperatoria.
Evaluación preoperatoria
La evaluación de la función cardíaca debe ser integral, considerando la presencia de comorbilidades, la capacidad funcional, desencadenantes de síntomas, fármacos de uso crónico, último control con electro fisiólogo y laboratorio cardiológico esencial.
El manejo de la ansiedad prequirúrgica debe ser considerado, aunque no existen recomendaciones basadas en la evidencia al respecto. Fisiopatológicamente, la taquicardia y la hiperadrenergia podrían desencadenar arritmias ventriculares graves. Por ello, se ha optado en diferentes series de casos por premedicar con ansiolíticos, siendo las benzodiacepinas, opioides y agonistas a-2 opciones seguras[7],[11],[16],[17].
Es fundamental mantener la medicación antiarrítmica y los P-bloqueadores en el período preoperatorio. Se recomienda que el paciente reciba su tratamiento habitual el día de la cirugía.
-
Laboratorio cardiológico esencial
Un electrocardiograma (ECG) de 12 derivaciones resulta fundamental, ya que solo el 12% de los casos presentan ECG normal[12]. Siendo característico de la enfermedad la presencia de onda épsilon, puede ser acompañado además de extrasístoles ventriculares, bloqueos de ramas. Alteraciones de progresión de onda R en derivaciones precordiales, asociado a ondas T negativas más allá de V1-V3 son asociados a patología avanzada, y compromiso bi-ventricular[8],[18].
La importancia de tener un trazado basal, radica en que se han reportado cambios en el ECG previos al desarrollo de arritmias ventriculares, incluyendo fibrilación auricular de novo, bloqueo auriculoventricular y extrasístoles ventriculares[18].
Un ecocardiograma es clave para evaluar la función sistólica y diastólica de ambos ventrículos, el nivel de infiltración miocárdica y las alteraciones segmentarias, así como el compromiso valvular secundario.

En pacientes portadores de DCI, es esencial coordinar entre los equipos de electrofisiología, cirugía y anestesia para definir el plan quirúrgico, controlar la función del DCI, realizar reprogramaciones preoperatorias y asegurar la monitorización intraoperatoria adecuada[19].
-
Manejo intraoperatorio
El objetivo principal del manejo intraoperatorio es evitar los factores desencadenantes de eventos arritmogénicos y mantener un gasto cardíaco adecuado.
Monitorización
No existe consenso sobre los estándares de monitorización en estos pacientes, pero se recomienda cumplir con los mínimos establecidos por la Sociedad Americana de Anestesiología (ASA). Además, se sugiere el uso de monitorización invasiva con línea arterial ante cualquier indicio de compromiso hemodinámico. En procedimientos de mayor envergadura, podría ser necesario utilizar ecocardiografía transesofágica para evaluar continuamente la función cardíaca.
Anestésicos locales
La lidocaína es segura, mientras que la bupivacaina, debido a su perfil cardiotóxico, debe administrarse en dosis más bajas. En caso de utilizarse en anestesia regional, no debe combinarse con epinefrina[17].
Antieméticos
La dexametasona como profilaxis no presenta contraindicaciones. Sin embargo, los antagonistas de 5-HT3 como el ondansetrón y los antagonistas D2 como la metoclopramida deben usarse con precaución debido a su posible efecto arritmogénico[20],[21].
Inductores y relajantes musculares
Los inductores anestésicos habituales, como las benzodiacepinas, propofol, etomidato y opioides, son seguros en estos pacientes el pancuronio podría estar contraindicado debido a su capacidad para aumentar la frecuencia cardíaca, aunque ha sido utilizado de manera segura en varios casos. El rocuronio no presenta contraindicaciones. Aunque los pacientes no presentan riesgo de hipertermia maligna, se debe evitar la succinilcolina[22].
Mantención anestesica
Los halogenados como el isoflurano y el sevoflurano son opciones seguras, y el uso de infusión de propofol con opioides también ha sido reportado como efectivo[16],[23].
Un estudio de Dong et al., sugiere que mantener la profundidad anestésica con un índice biespectral (BIS) menor de 40 podría tener un efecto protector contra las taquicardias ventriculares[24].
Drogas vasoactivas
No se recomienda el uso de fármacos pro-arritmogénicos como la dobutamina o la epinefrina, aunque no hay evidencia sólida que respalde esta contraindicación. La dopamina y la noradrenalina se han utilizado de manera segura en algunas series de casos[16],[23].
-
Cirugía obstétrica
El manejo de pacientes con DAVD durante el embarazo y el parto ha recibido más atención en los últimos años, aunque la información disponible es limitada. Se ha reportado que los cambios hemodinámicos durante el embarazo son bien tolerados en estos pacientes[25]-[27].
Se ha logrado realizar con éxito partos vaginales con analgesia peridural y cesáreas con anestesia peridural sola o combinada con anestesia general[25]-[28].
-
Posoperatorio
Dado que se han reportado arritmias graves en el posoperatorio[29], se recomienda monitorización en una unidad de cuidados intensivos durante las primeras 24 h. La reprogramación del DCI después de la cirugía es fundamental, por lo que debe haber una coordinación adecuada entre los equipos para un seguimiento óptimo[19].
Es esencial también garantizar un adecuado manejo del dolor mediante un esquema multimodal para prevenir la hiperadrenergia secundaria al dolor no tratado.
-
Conclusiones
Dado que la DAVD es una patología rara en América Latina, es crucial su conocimiento debido a su asociación con arritmias potencialmente letales y las complicaciones cardiovasculares que pueden surgir durante procedimientos quirúrgicos. Una comprensión profunda de esta enfermedad permite realizar una planificación perioperatoria adecuada, optimizando la seguridad del paciente mediante estrategias dirigidas a minimizar los desencadenantes de arritmias.
-
Referencias
1. Marcus FI, Fontaine GH, Guiraudon G, Frank R, Laurenceau JL, Malergue C, et al. Right Ventricular Dysplasia: A Report of 24 Adult Cases [Internet]. Available from: http://ahajournals.org
2. Te Riele AS, Hauer RN. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Clinical challenges in a changing disease spectrum. Vol. 25, Trends in Cardiovascular Medicine. Elsevier Inc.; 2015. p. 191–8.
3. Corrado D, Fontaine G, Marcus FI, Mckenna WJ, Nava A, Thiene G, et al. Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Need for an International Registry Morbid Anatomy and Histology Current Perspective [Internet]. 2000. Available from: http://www.circulationaha.org
4. Staikou C, Chondrogiannis K, Mani A. Perioperative management of hereditary arrhythmogenic syndromes. Vol. 108, British Journal of Anaesthesia. Oxford University Press; 2012. p. 730–44. https://doi.org/10.1093/bja/aes105.
5. Protonotarios N, Tsatsopoulou A. Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Pathol. 2004;13(4):185–94. https://doi.org/10.1016/j.carpath.2004.03.609 PMID:15210133
6. Karmouch J, Protonotarios A, Syrris P. Genetic basis of arrhythmogenic cardiomyopathy. Vol. 33, Current Opinion in Cardiology. Lippincott Williams and Wilkins; 2018. p. 276–81. https://doi.org/10.1097/HCO.0000000000000509.
7. Roudijk RW, Verheul L, Bosman LP, Bourfiss M, Breur JM, Slieker MG, et al. Clinical Characteristics and Follow-Up of Pediatric-Onset Arrhythmogenic Right Ventricular Cardiomyopathy. JACC Clin Electrophysiol. 2022 Mar;8(3):306–18. https://doi.org/10.1016/j.jacep.2021.09.001 PMID:35331425
8. Corrado D, Link MS, Calkins H. Arrhythmogenic Right Ventricular Cardiomyopathy. Jarcho JA, editor. New England Journal of Medicine [Internet]. 2017 Jan 5;376(1):61–72. Available from: http://www.nejm.org/doi/10.1056/NEJMra1509267 https://doi.org/10.1056/NEJMra1509267.
9. Fontaine G, Frank R, Tonet JL, Guiraudon G, Cabrol C, Chomette G, et al. Arrhythmogenic right ventricular dysplasia: a clinical model for the study of chronic ventricular tachycardia. Jpn Circ J. 1984 Jun;48(6):515–38. https://doi.org/10.1253/jcj.48.515 PMID:6376841
10. Neto JE, Tonet J, Frank R, Fontaine G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D)-what we have learned after 40 years of the diagnosis of this clinical entity. Vol. 112, Arquivos Brasileiros de Cardiologia. Arq Bras Cardiol. 2019;•••:91–103.
11. Yildiz H, Silay E, Coskuner I, Ozyurt K, Olgar S, Senoglu N, et al. Anaesthesia in Naxos disease: first case report. Bosn J Basic Med Sci. 2013 Feb;13(1):63–5. https://doi.org/10.17305/bjbms.2013.2421 PMID:23448613
12. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010 Apr;121(13):1533–41. https://doi.org/10.1161/CIRCULATIONAHA.108.840827 PMID:20172911
13. Corrado D, Anastasakis A, Basso C, Bauce B, Blomström-Lundqvist C, Bucciarelli-Ducci C, et al. Proposed diagnostic criteria for arrhythmogenic cardiomyopathy: european Task Force consensus report. Int J Cardiol. 2024 Jan;395:131447. https://doi.org/10.1016/j.ijcard.2023.131447 PMID:37844667
14. Corrado D, Wichter T, Link MS, Hauer RN, Marchlinski FE, Anastasakis A, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015 Aug;132(5):441–53. https://doi.org/10.1161/CIRCULATIONAHA.115.017944 PMID:26216213
15. Toh KW, Nadesan K, Sie MY, Vijeyasingam R, Tan PS. Postoperative death in a patient with unrecognized arrhythmogenic right ventricular dysplasia syndrome. Anesth Analg. 2004 Aug;99(2):350–2. https://doi.org/10.1213/01.ANE.0000132996.12248.B0 PMID:15271703
16. Motta P, Mossad E, Savage R. Right ventricular exclusion surgery for arrhythmogenic right ventricular dysplasia with cardiomyopathy. Anesth Analg. 2003 Jun;96(6):1598–602. https://doi.org/10.1213/01.ANE.0000060452.30003.39 PMID:12760981
17. Levy D, Bigham C, Tomlinson D. Anaesthesia for patients with hereditary arrhythmias; part 2: congenital long QT syndrome and arrhythmogenic right ventricular cardiomyopathy. Vol. 18, BJA Education. Elsevier Ltd; 2018. p. 246–53.
18. Zhang L, Liu L, Kowey PR, Fontaine GH. The electrocardiographic manifestations of arrhythmogenic right ventricular dysplasia. Curr Cardiol Rev. 2014 Aug;10(3):237–45. https://doi.org/10.2174/1573403X10666140514102928 PMID:24827798
19. Wan EY, Rogers AJ, Lavelle M, Marcus M, Stone SA, Ottoboni L, et al. Periprocedural Management and Multidisciplinary Care Pathways for Patients With Cardiac Implantable Electronic Devices: A Scientific Statement From the American Heart Association. Vol. 150, Circulation. Lippincott Williams and Wilkins; 2024. p. e183–96.
20. Baguley WA, Hay WT, Mackie KP, Cheney FW, Cullen BF. Cardiac Dysrhythmias Associated with the Intravenous Administration of Ondansetron and Metoclopramide [Internet]. Vol. 84, Anesth Analg. 1997. Available from: http://journals.lww.com/anesthesia-analgesia
21. Afonso N, Dang A, Namshikar V, Kamat S, Rataboli PV. Intravenous ondansetron causing severe bradycardia: two cases. Ann Card Anaesth. 2009;12(2):172–3. PMID:19602754
22. Swan H, Laitinen PJ, Toivonen L. Volatile anesthetics and succinylcholine in cardiac ryanodine receptor defects. Anesth Analg. 2004 Aug;99(2):435–7. https://doi.org/10.1213/01.ANE.0000130395.93107.15 PMID:15271719
23. Valchanov K, Goddard M, Ghosh S. Anesthesia for heart transplantation in patients with arrhythmogenic right ventricular dysplasia. J Cardiothorac Vasc Anesth. 2014 Apr;28(2):355–7. https://doi.org/10.1053/j.jvca.2013.02.025 PMID:23994174
24. Dong H, Li N, Sun Z. The effect of anesthesia depth on radiofrequency catheter ablation of ventricular tachycardia: a retrospective study. BMC Anesthesiol. 2021 Nov;21(1):285. https://doi.org/10.1186/s12871-021-01503-6 PMID:34781892
25. Doyle NM, Monga M, Montgomery B, Dougherty AH. Arrhythmogenic right ventricular cardiomyopathy with implantable cardioverter defibrillator placement in pregnancy. J Matern Fetal Neonatal Med. 2005 Aug;18(2):141–4. https://doi.org/10.1080/14767050500226500 PMID:16203602
26. Güdücü N, Kutay SS, Ozenç E, Ciftçi C, Yiğiter AB, Işçi H. Management of a rare case of arrhythmogenic right ventricular dysplasia in pregnancy: a case report. J Med Case Rep. 2011 Jul;5(1):300. https://doi.org/10.1186/1752-1947-5-300 PMID:21740597
27. Bauce B, Daliento L, Frigo G, Russo G, Nava A. Pregnancy in women with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Obstet Gynecol Reprod Biol. 2006 Aug;127(2):186–9. https://doi.org/10.1016/j.ejogrb.2005.10.011 PMID:16337730
28. Iriyama T, Kamei Y, Kozuma S, Taketani Y. Management of patient with arrhythmogenic right ventricular cardiomyopathy during pregnancy. J Obstet Gynaecol Res. 2013 Jan;39(1):390–4. https://doi.org/10.1111/j.1447-0756.2012.01954.x PMID:22889369
29. Tabib A, Loire R, Miras A, Thivolet-Bejui F, Timour Q, Bui-Xuan B, et al. Unsuspected cardiac lesions associated with sudden unexpected perioperative death. European Journal of Anaesthesiology | EJA [Internet]. 2000;17(4). Available from: https://journals.lww.com/ejanaesthesiology/fulltext/2000/04000/unsuspected_cardiac_lesions_associated_with_sudden.4.aspx https://doi.org/10.1097/00003643-200004000-00004.