Manuel Enrique Rodríguez García1*, Yassel Arias Otamendy2, Annia Aguirre Ruiz2
 ORCID
ORCIDRecibido: 11-02-2025
Aceptado: 14-04-2025
©2025 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 54 Núm. 5 pp. 748-751|https://doi.org/10.25237/revchilanestv54n5-37
PDF|ePub|RIS
Diagnostic and therapeutic challenges of refractory cardiogenic shock in a pediatric patient with J-wave electrocardiographic pattern. Case report
Abstract
The J-wave syndrome encompasses disorders such as Brugada syndrome (BrS) and early repolarization syndrome (ERS), both associated with ionic abnormalities in cellular repolarization. These syndromes can coexist with short QT syndrome (SQTS), characterized by shortened corrected QT intervals. Cardiogenic shock in these patients is difficult to manage, with limited response to inotropes and vasopressors. This case report presents a child with cardiogenic shock secondary to a complex repolarization disorder (CRD), where ionic channel abnormalities affect contractility and response to conventional treatments.
Resumen
El síndrome de onda J abarca trastornos como el síndrome de Brugada (BrS) y el síndrome de repolarización precoz (ERS), ambos asociados con alteraciones iónicas en la repolarización celular. Estos síndromes pueden coexistir con el síndrome de QT corto (SQTS), caracterizado por intervalos QT corregidos abreviados. El shock cardiogénico en estos pacientes es difícil de tratar, con respuesta limitada a inotrópicos y vasopresores. Se presenta el caso de un niño con shock cardiogénico secundario a un trastorno de repolarización complejo (TRC), donde las alteraciones en los canales iónicos afectan la contractilidad y la respuesta a tratamientos convencionales.
-
Introducción
Los síndromes de onda J son canalopatías hereditarias que incluyen el síndrome de Brugada (BrS) y el síndrome de repolarización precoz (ERS), caracterizados por la presencia de ondas J acentuadas. En BrS, estas ondas se manifiestan como una elevación cóncava del segmento ST en las derivaciones electrocardiográficas precordiales derechas (V1 – V3). En el caso de ERS, se presenta una onda J distintiva o una elevación del punto J en dos derivaciones electrocardiográficas contiguas inferolaterales (I, II, III, aVL, aVF, V4 – V6).
Los mecanismos iónicos y celulares subyacentes al BrS y al ERS están vinculados a alteraciones en la repolarización celular debido a un gradiente transmembrana anómalo, causado por una distribución desigual de las corrientes iónicas. En BrS, la disfunción de los canales de sodio (INa) y un aumento de la corriente transitoria de potasio (Ito) en el tracto de salida del ventrículo derecho (RVOT) provocan un cambio en el potencial de acción, generando un “notch”. En ERS, este fenómeno se produce en el ventrículo izquierdo (LV), y está asociado principalmente con un aumento de Ito[1].
Los síndromes de onda J se presentan con un intervalo QT relativamente corto en pacientes portadores de mutaciones en genes de canales de calcio[2]. Sin embargo, pueden coexistir con el síndrome de QT corto (SQTS) por ganancia de función de los canales de potasio, ambos trastornos se caracterizan por intervalos QT corregidos abreviados en el electrocardiograma[3] aunque difieren en su perfil pronóstico.
A su vez, los canales de K+ juegan un papel crucial en la repolarización del potencial de acción de la célula cardiaca[4], mutaciones en los genes que codifican estos canales pueden provocar aumento en la densidad de KvLQT1 (Canales de potasio tipo I-Ks)[5], lo que acorta la duración del potencial de acción (DPA) y genera SQTS, con reducción de la eficiencia del bombeo ventricular superior al 60%[4]. Esta pérdida de eficiencia podría acompañarse de signos clínicos de shock en ausencia de arritmias cardíacas graves forma de presentación más frecuente en estos casos.
El shock cardiogénico es una emergencia médica cuya respuesta a tratamientos convencionales, como inotrópicos y vasopresores, puede ser limitada o incluso nula en pacientes con Trastornos de Repolarización Complejos (TRC). Esta condición subraya la necesidad de un enfoque multidisciplinario altamente especializado que permita comprender los mecanismos subyacentes de esta presentación clínica y, así, mejorar los resultados en estos pacientes.
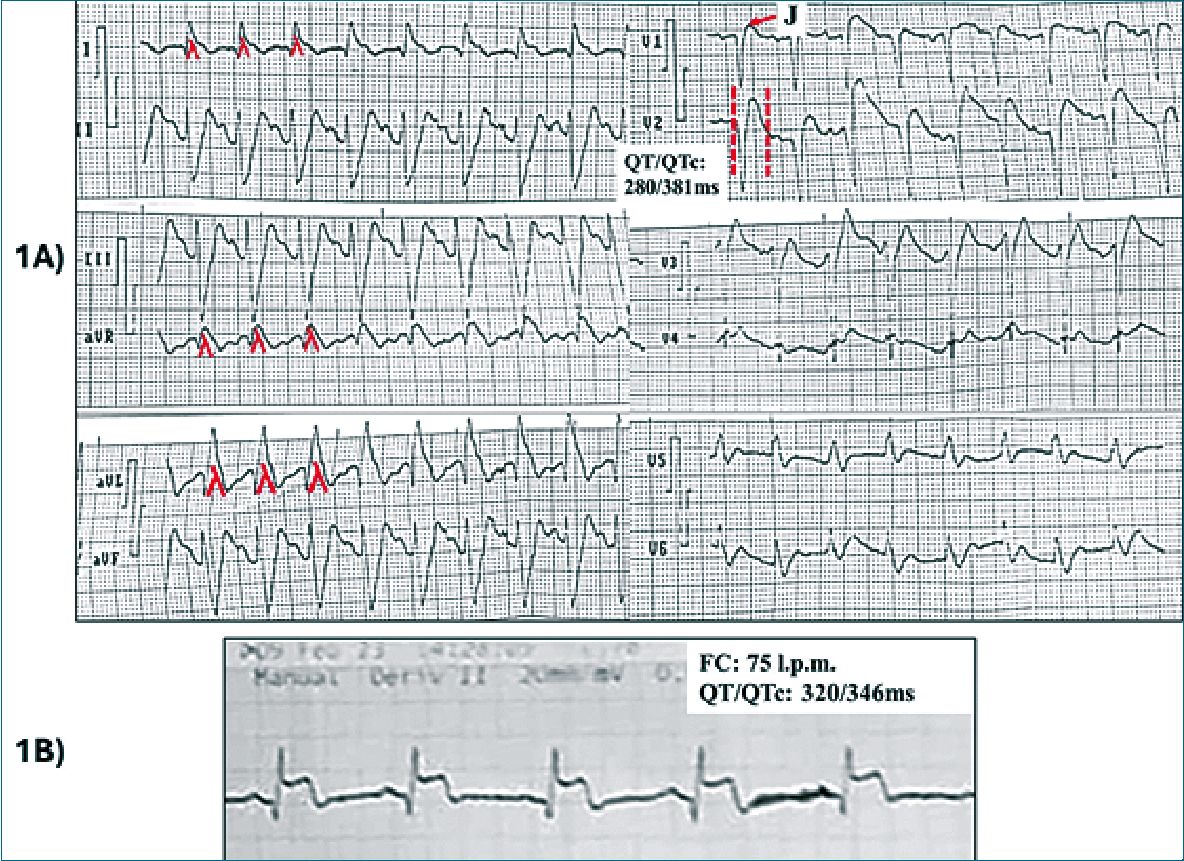
Figura 1. A) ECG de 12 derivaciones. Taquicardia sinusal 125 lpm, intervalo PR 150 ms, QRS duración: 120 ms, eje QRS: – 20 grados (desviación axial izquierda). Se observa la onda Lambda u onda de Gussak, caracterizada por una elevación del ST de perfil triangular o de convexidad superior > o = 2 mm seguido de onda T negativa en D1, aVL y aVR, con imagen recíproca en espejo en pared inferior. Patrón de Brugada tipo 2 en V1- V2. Intervalo QT/QTc (Rautaharju): 280/381ms. Signo J-Tpico: 80-100 ms; 1B) DII largo, ritmo sinusal, FC: 75 lpm. Elevación del segmento ST > 2 mm. QT/QTc (Rautaharju): 320/346 ms. QT rígido, escasa variabilidad con la Frecuencia cardíaca.
-
Reporte de caso
Se presenta el caso de un paciente masculino de 12 años, ingresado en la Unidad de Cuidados Intensivos Emergentes por agitación psicomotora y obnubilación súbita, con un episodio similar tres años antes y sin secuelas. Entre sus antecedentes heredofamiliares destaca la pérdida de un hermano menor, a los 4 años, por tetralogía de Fallot.
A su ingreso presentó signos de inestabilidad hemodinámica: sudoración profusa, frialdad, cianosis distal, ausencia de pulso periférico y tensión arterial, y pupilas isocóricas y reactivas. El electrocardiograma (ECG) mostró taquicardia sinusal, síndrome de onda J asociado a SQTS (Figuras 1A y 1B). El ultrasonido a pie de cama (POCUS) mostro función contráctil ventricular conservada, signos de edema pulmonar y ausencia de líquido libre abdominal.
Exámenes de laboratorio:
• Hemoglobina: 124 g/L.
• Leucocitos: 11,5 x 109/L.
• Glicemia: 5,7 mmol/L.
• Gasometría: acidosis metabólica con Gap elevado.
• Ionograma: normal.
• Estudios toxicológicos: negativos.
Diagnóstico: Shock cardiogénico, basado en el perfil hemo- dinámico y el contexto clínico.
Se inició manejo avanzado de la vía aérea con ventilación mecánica controlada y monitorización continua. Se administraron 500 ml de solución salina al 0,9%, lo que provocó edema pulmonar cardiogénico y se suspendió la fluidoterapia. Se inició soporte con vasopresores (noradrenalina 0,3-3 mcg/kg/min), observándose elevación del segmento ST, que regresó a la línea base tras reducir la dosis de noradrenalina. No se logró mejorar la tensión arterial ni restaurar los pulsos periféricos, por lo que se administró dobutamina (10-20 mcg/kg/min) sin respuesta. A los 30 minutos, el paciente sufrió parada cardiaca en ritmo idioventricular, iniciándose reanimación cardiopulmonar con epinefrina cada 3 minutos. Después de 15 minutos, se obtuvo ritmo sinusal, pero sin tensión arterial ni pulso periférico. Este ritmo se mantuvo menos de cinco minutos, a pesar del soporte inotrópico y vasopresor. Posteriormente, el paciente volvió a ritmo idioventricular, que degeneró en asistolia, sin respuesta a la reanimación.
Hallazgos en la necropsia:
• Edema pulmonar.
• Edema cerebral.
-
Discusión
El shock cardiogénico es una condición en la cual el corazón no puede generar suficiente gasto cardíaco para satisfacer las demandas del organismo[6]. En este caso se asoció a TRC, que involucra la superposición del síndrome de onda J, incluido el patrón de Brugada y el síndrome QT corto. Estos trastornos, pueden inducir alteraciones en los discos intercalares (IDs), que son esenciales para la adherencia celular y la conducción eléctrica cardíaca.
El connexoma, que agrupa los componentes celulares del IDs, regula tanto la estabilidad mecánica como la propagación de señales eléctricas[7].
En casos de shock cardiogénico relacionado con TRC, alteraciones genéticas en el connexoma pueden afectar la adherencia celular y la conducción eléctrica, contribuyendo a la disfunción miocárdica.
Aunque la onda J no es un marcador directo de disfunción contráctil en términos de fracción de eyección o movimientos de las paredes ventriculares, sí refleja alteraciones eléctricas subyacentes que afectan la contracción y relajación del miocardio[1].
En el síndrome de Brugada, la expresión de la proteína con- nexina-43 (C-43) está anormalmente disminuida en el tracto de salida del ventrículo derecho, lo que altera la repolarización y despolarización[8]. Además, algunos pacientes con patrón de Brugada, presentan mutaciones en genes que codifican canales de potasio dependientes de voltaje, como KCNE (subunidades accesorias de los canales IKs y Ito), KCNE3 (IKs)[7].
En relación con lo anterior, el shock cardiogénico puede surgir debido a una ganancia de función de los canales de potasio transitorios (Ito), lo que compromete la excitabilidad y la contractilidad miocárdica secundario a desajustes en el acoplamiento excitación-contracción.
La presencia de síndrome QT corto (SQTS) y síndrome de onda J sugiere múltiples variantes genéticas responsables de la disfunción miocárdica y vascular, lo cual está respaldado por, el mayor estudio genómico sobre el intervalo QT realizado en 75.000 individuos, identificó varios SNPs (polimorfismos de nucleótido único) que explican el 10% de la variación del QT, lo que sugiere una base genética poligénica[9].
El diagnóstico del síndrome de QT corto (SQTS) mediante mutaciones genéticas tiene una tasa de éxito inferior al 30%, sin embargo, más del 80% de los pacientes genotipados presentan mutaciones en los canales IKr y IKs, que son los principales involucrados en la condición[3].
Las alteraciones en los canales de potasio fundamentales para la despolarización y repolarización cardíaca, pueden ocasionar disfunción contráctil o de relajación por cambios en la energía de plegamiento y movilidad miocárdica, los cuales no siempre se reflejan en el ecocardiograma[10].
De hecho, se describen mecanismos moleculares e iónicos en el desajuste contracción-relajación debido a ganancia de función Canales KvLQT1; cuanto mayor es la conductividad de la membrana en el canal de K+, más corto es (APD) lo que reduce las concentraciones de calcio (Ca2+) intracelular por acortamiento del tiempo de apertura del canal de Ca2+ tipo L.
La disminución de la APD está relacionada con disminución de la fuerza de contracción y consumo de ATP del músculo en el momento telesistólico; lo que incrementa la tensión de los ventrículos en el momento telediastólico. Estas alteraciones fisiológicas se acompañan de disminución de la presión en el ventrículo izquierdo, aumento del volumen telediastólico del ventrículo izquierdo, así como una reducción de la presión del pulso y en las arterias sistémicas durante el ritmo sinusal[4].
Las alteraciones en la propagación del potencial de acción reducen la respuesta a los inotrópicos cuya acción farmacológica es incrementar la contractilidad al aumentar el flujo de calcio hacia las células del miocardio. Esto se ve agravado por la disfunción diastólica, la cual no puede ser corregida por inotrópicos.
La actividad de los canales de potasio regula paralelamente el tono vascular, al afectar el potencial de membrana de las células del músculo liso vascular. La apertura de estos canales provoca vasodilatación al reducir la entrada de calcio. Los canales de potasio activados por calcio, de gran conductancia y despolarización, regulan el tono y diámetro vascular, contribuyendo así a la vasodilatación.
La coexistencia de mutaciones en los canales de potasio puede agravar las condiciones cardiovasculares, lo cual parece guardar relación con desregulaciones del sistema nervioso autónomo que alteran la respuesta a las catecolaminas y reducen la efectividad de los vasopresores[1].
La complejidad de estas alteraciones fisiopatológicas explica el fracaso circulatorio agudo total con desarrollo de edema pulmonar cardiogénico severo y la ausencia de respuesta a los inotrópicos y vasopresores, en el que, la utilización de un dispositivo de asistencia circulatoria habría sido la medida de rescate indicada.
Las canalopatías tienen un impacto significativo en la estructura y función miocárdica, afectando la contracción y relajación celular[1], alteraciones que no siempre se reflejan en el ecocardiograma convencional. La disfunción diastólica, caracterizada por una relajación incompleta, suele ser subclínica y difícil de detectar, pero puede ser crucial en el desarrollo del shock cardiogénico, incluso sin cambios en la fracción de eyección o la geometría cardíaca.
-
Conclusiones
La presentación de shock cardiogénico asociado a un Trastorno de Repolarización Complejo (TRC), caracterizado por la superposición de síndrome de onda J y síndrome de QT corto, resalta la implicación de disfunciones en los canales iónicos. Estas alteraciones afectan tanto la repolarización celular como la contractilidad miocárdica y el tono vascular. Además, el compromiso del connexoma, responsable de regular la adherencia celular y la conducción eléctrica, puede dificultar la respuesta a inotrópicos y vasopresores, lo que subraya la necesidad de un enfoque diagnostico – terapéutico especializado.
Confidencialidad de la información: se han cumplido con los protocolos de nuestro centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informado: Los autores hemos obtenido el consentimiento informado por escrito del padre del niño fallecido mencionados en el artículo. El autor para correspondencia está en posesión del presente documento.
MERG: reunió la información, aportó datos o herramientas de análisis, escribió el documento.
YAO y ACR: recabaron los datos, aportaron información o herramientas analíticas.
Apoyo financiero y patrocinio:
No se recibió apoyo económico ni financiamiento.
Conflicto de intereses:
Ninguno.
-
Referencias
1. Antzelevitch C, Di Diego J. J wave syndromes: what’s new? Trends Cardiovasc Med. 2021;31(1):1–9. https://doi.org/10.1016/j.tcm.2021.07.001 PMID:34256120
2. Belenkov Y, Snezhitskiy V, Gizatulina T, Shpak NV, Kuznetsov VA, Martyanova LU, et al. Update of the diagnostic criteria of J-wave syndrome: new concepts and their relevance to cardiology practice. Kardiologiia. 2018;58(11):41–52. https://doi.org/10.18087/cardio.2018.11.10196 PMID:30625077
3. Hancox JC, Du CY, Butler A, Zhang Y, Dempsey CE, Harmer SC, et al. Pro-arrhythmic effects of gain-of-function potassium channel mutations in the short QT syndrome. Philos Trans R Soc Lond B Biol Sci. 2023 Jun;378(1879):20220165. https://doi.org/10.1098/rstb.2022.0165 PMID:37122211
4. Jeong DU, Lim KM. The effect of myocardial action potential duration on cardiac pumping efficacy: a computational study. Biomed Eng Online. 2018 Jun;17(1):79. https://doi.org/10.1186/s12938-018-0508-2 PMID:29907152
5. Issa Z, Miller J, Zipes D. Clinical Arrhythmology and Electrophysiology: A Companion to Braunwald’s Heart Disease. 4th ed. Philadelphia: Elsevier; 2023. pp. 1046–117. ISBN: 978-0-323-88182-1.
6. Othman H, Hamzah M, Aly H. Outcomes of cardiogenic shock in pediatric cardiomyopathy versus congenital heart disease: Analysis of the National Inpatient Sample Database. J Am Coll Cardiol. 2020;75(6):854. https://doi.org/10.1016/S0735-1097(20)31481-9.
7. Ben-Haim Y, Asimaki A, Behr ER. Brugada syndrome and arrhythmogenic cardiomyopathy: overlapping disorders of the connexome? Europace. 2021 May;23(5):653–64. https://doi.org/10.1093/europace/euaa277 PMID:33200179
8. Rodríguez-Sinovas A, Sánchez JA, Valls-Lacalle L, Consegal M, Ferreira-González I. Connexins in the heart: Regulation, function and involvement in cardiac disease. Int J Mol Sci. 2021 Apr;22(9):4413. https://doi.org/10.3390/ijms22094413 PMID:33922534
9. Cerrone M, Remme CA, Tadros R, Bezzina CR, Delmar M. Beyond the one gene-one disease paradigm. Circulation. 2019 Aug;140(7):595–610. https://doi.org/10.1161/CIRCULATIONAHA.118.035954 PMID:31403841
10. Agudelo WA, Gil-Quiñones SR, Fonseca A, Arenas A, Castro L, Sierra-Díaz DC, et al. Structural modelling of KCNQ1 and KCNH2 double mutant proteins, identified in two severe long QT syndrome cases, reveals new insights into cardiac channelopathies. Int J Mol Sci. 2021 Nov;22(23):12861. https://doi.org/10.3390/ijms222312861 PMID:34884666