
Javiera Valeria Gallegos 1 , Cindy Raddatz Martínez 1
 ORCID
ORCIDRecibido: 17-11-2020
Aceptado: 26-11-2020
©2021 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 50 Núm. 1 pp. 79-89|https://doi.org/10.25237/revchilanestv50n01-06
PDF|ePub|RIS
Malignant huperthermia
Abstract
Carriers of malignant hyperthermia (MH), a subclinical metabolic myopathy, respond differently to the general population in response to triggering agents, such as volatile anesthetics and succinylcholine. Its incidence ranges from 1:10,000 to 1:250,000 anesthesias. Using etiological treatment, the current mortality rate is around 5%. The biochemical, metabolic, and physiological deterioration generally associated with MH is a direct result of a sudden and progressive increase in intracellular calcium in striated muscle cells. This generates a hypermetabolic state, with a rapid increase in body temperature that can lead to a fatal outcome if not diagnosed and treated in time. The genetic factors that determine susceptibility to MH are complex, with the participation of more than one gene. Its clinical symptoms are highly variable, from mild or moderate to fulminant attacks with severe muscle hypermetabolism and rhabdomyolysis. Capnography and pulse oximetry have great clinical diagnostic value. Other early symptoms of an MH attack may include sinus tachycardia, supraventricular or ventricular arrhythmia, and isolated masterean spasm or generalized muscle stiffness. The rise in temperature is a late sign. After an attack, or in possibly susceptible patients, the laboratory diagnosis is made with the in vitro contracture test, in which the contraction of muscle fibers, obtained through a skeletal muscle biopsy, is studied in the presence of halothane or caffeine. In patients known to be susceptible to MH, neuraxial and regional techniques should be preferred if surgery allows it; otherwise, trigger-free anesthetic methods (TIVA) should be available. Management of the MH crisis is based on three main actions: 1) stopping the administration of halogenates; 2) hyperventilation with 100% oxygen, and 3) administration of intravenous dantrolene.
Resumen
La hipertermia maligna (HM) es una miopatía metabólica subclínica, cuyos portadores tienen una respuesta diferente a la población general ante la presencia de un agente desencadenante: anestésicos volátiles y succinilcolina. Su incidencia tiene rangos entre 1:10.000 a 1:250.000 anestesias. Su mortalidad actual usando tratamiento etiológico es de 5%. El deterioro bioquímico, metabólico y fisiológico asociado clásicamente al cuadro de HM es el resultado directo de un aumento súbito y progresivo del calcio intracelular de las células musculares estriadas, que genera un estado hipermetabólico, calor y un rápido aumento de la temperatura corporal, que puede llevar a un desenlace fatal si no se diagnostica y se trata a tiempo. Su herencia es complicada: se trata de una transmisión multigénica, en que la susceptibilidad a la HM depende de más de un gen. Los síntomas clínicos son muy variables, desde leves o moderados hasta crisis fulminantes con hipermetabolismo muscular severo y rabdomiólisis. La capnografía y la oximetría tiene un gran valor diagnóstico clínico. Otros síntomas tempranos de una crisis de HM pueden incluir taquicardia sinusal, arritmia supraventricular o ventricular y espasmo de maséteros aislado o rigidez muscular generalizada; el aumento de la temperatura es un signo tardío. Después de la crisis o en los pacientes posiblemente susceptibles, el diagnóstico de laboratorio se hace con el test de contractura , basado en la contracción de fibras musculares tomadas a partir de una biopsia de músculo estriado en presencia de halotano o cafeína. En los pacientes conocidamente susceptibles a HM se debe preferir las técnicas neuroaxiales y regionales si la cirugía lo permite; en caso contrario, debe disponerse de métodos anestésicos libres de agentes desencadenantes (TIVA). El manejo de la crisis de HM está basado en tres medidas principales: 1) la detención de la administración de halogenados; 2) la hiperventilación con oxígeno al 100% y 3) la administración de dantrolene endovenoso.
-
Introducción
La hipertermia maligna (HM) es un trastorno muscular que se hereda en forma autosómica dominante y se manifiesta por una respuesta hipermetabólica, con una liberación exagerada de calcio del retículo sarcoplásmico, que puede ser desencadenado en el perioperatorio por anestésicos volátiles (halotano, isoflurano, sevoflurano y desflurano) y bloqueadores neuromusculares despolarizantes (succinilcolina). Desde el punto de vista clínico es una miopatía metabólica subclínica, cuyos portadores, llamados susceptibles a HM (MHS por sus siglas en inglés), tienen una respuesta diferente a la población general ante la presencia de un agente desencadenante. Se describe mejor como un síndrome, pues no todos los pacientes tienen la misma respuesta cada vez que son expuestos a un agente desencadenante.
El 8 de noviembre de 1981 ocurrió en la Asistencia Pública el primer episodio de hipertermia maligna diagnosticado clínicamente. Fue publicado en un artículo de la Revista de la Asistencia Pública[1]. Posteriormente, hubo 3 casos y luego una sequía de más de 15 años en que no ocurrió ninguno. En los últimos 5 años hubo una serie clínica de 3 casos. En el primer caso el tratamiento fue sólo sintomático; los dos casos siguientes se trataron con una solución de dantrolene obtenida a partir de cápsulas para uso oral y en todos los siguientes se utilizó la droga original liofilizada. La secuencia descrita da cuenta de dos datos relevantes: por un lado, la variabilidad con que se puede presentar casos a lo largo del tiempo en una institución, por el otro, el avance en la disposición de una terapia efectiva.
-
Epidemiología
Es difícil determinar la incidencia de los episodios de HM porque hay muchas formas clínicas. Puesto que se trata de una enfermedad subclínica, su incidencia depende de múltiples factores, como la posibilidad de comprobar el diagnóstico, la exposición a un agente desencadenante por un tiempo suficiente y otra serie de factores (lugar geográfico estudiado, edad, sexo, raza, factores ambientales, etc.). Aunque no es una enfermedad exclusiva de un grupo racial, el grupo más afectado es el caucásico, especialmente originario del norte de Europa.
Es así, que tiene una incidencia variable, con grandes fluctuaciones en los reportes publicados con rangos entre 1:10.000 a 1:250.000 anestesias[2]. El 75% corresponde a hombres con un promedio de edad de 22 años, aunque también hay reporte de casos en ancianos. Los episodios agudos son más frecuentes en hombres, en pacientes con gran masa muscular o que han realizado recientemente ejercicio. En nuestro país se desconoce su incidencia.
Más del 50% de todas las reacciones de HM involucran a la población pediátrica y menores de 18 años. Aunque es un antecedente que se pregunta a todos los pacientes, sólo 6,5% refiere historia familiar de HM. Si bien se puede desarrollar una crisis de HM en la primera exposición a una anestesia, más de la mitad de los pacientes ha recibido más de 2 veces anestesia general previo a su presentación[3].
La prevalencia estimada de anomalías genéticas asociadas con la susceptibilidad a la HM puede ser tan grande como uno de cada 3.000 individuos (rango 1:3.000 a 1:8.500), con una estimación más reciente de 1 en 400.
Actualmente, la mortalidad en Estados Unidos de Norteamérica es menor al 5%.
-
Fisiopatología
El deterioro bioquímico, metabólico y fisiológico asociado clásicamente al cuadro de HM es el resultado directo de un aumento súbito y progresivo del calcio intracelular de las células musculares estriadas. El retículo sarcoplásmico es un sistema de membranas de la fibra muscular que está estructural y funcionalmente especializado en la rápida liberación y secuestro de los iones de Ca2+. Está ampliamente distribuido por todo el sarcoplasma rodeando las miofibrillas en forma de cisternas membranosas, túbulos y vesículas.
Las cisternas aplanadas del retículo sarcoplásmico, llamadas también cisternas terminales, están en contacto con una invaginación del sarcolema llamada túbulo transversal. El control de la liberación de Ca++ del músculo esquelético tiene lugar donde los túbulos transversales toman contacto con las cisternas terminales del retículo sarcoplásmico. El túbulo transversal y dos cisternas terminales adyacentes constituyen una tríada. Los canales de calcio implicados en la liberación del calcio del músculo son los receptores ryanodina y probablemente en mucho menor grado los receptores de la dihidropiridina (Figura 1).
En las personas normales, los niveles de Ca2+ en el mioplasma son controlados por el gen RyR1, por el DHPR y por el sistema Ca2+-adenosina trifosfatasa (Ca2+-ATPasa). En los pacientes susceptibles, durante una crisis de HM, el agente desencadenante induce la apertura prolongada de los receptores de ryanodina funcionalmente alterados, lo que produce una liberación excesiva de calcio del retículo sarcoplásmico y una contracción muscular mantenida que se presenta clínicamente como rigidez[4].
Además, la activación constante del metabolismo aeróbico y anaeróbico resulta en un mayor consumo de oxígeno, lo que conduce a hipoxia, acidosis láctica, producción excesiva de CO2 y aumento de la temperatura corporal. La recaptación de calcio en el retículo sarcoplásmico y la contracción muscular sostenida, consumen grandes cantidades de ATP. El agotamiento de las reservas celulares de ATP provoca hipoxia, acidosis metabólica y alteración en la integridad de la membrana; sobreviene rabdomiólisis (destrucción de las células musculares) y la consiguiente liberación del contenido de la célula (potasio, creatina fosfoquinasa, mioglobina, etc.) en la circulación[5].
La Figura 2 resume las alteraciones fisiopatológicas que ocurren en el retículo sarcoplásmico de la fibra muscular y que culminan con las manifestaciones clínicas de HM: hipercapnea, hipoxemia, acidosis, rigidez muscular, rabdomiólisis, hiperkalemia e hipertermia. Manifestaciones que si no se tratan oportunamente culminan con la muerte del paciente.
El estado hipermetabólico de la crisis de HM genera calor y a un rápido aumento de la temperatura corporal, que puede llevar a un desenlace fatal sino se diagnostica y se trata a tiempo.
-
Genética
Siempre se ha intentado sobresimplificar la transmisión genética de la susceptibilidad a la HM en los seres humanos, describiéndola como autosómica dominante, con una penetrancia incompleta. Cuando se trata de una transmisión autosómica dominante, el sujeto heterocigoto transmite la mutación a su descendencia con 50% de probabilidad. Sin embargo, el asunto no es tan simple y, a la luz de los conocimientos actuales, debe considerarse la influencia de un trasfondo genético común, que abarca alelos comunes en loci de diferentes susceptibilidades, cada uno de los cuales contribuye con efectos fenotípicos de menor importancia. Se trata de una transmisión multigénica, en que la susceptibilidad a la HM depende de más de un gen. Sorprendentemente los homocigotos no presentan sintomatología clínica ni positividad a los tests de contractura. Para complicar aún más las cosas, en algunas familias el modelo parece recesivo.
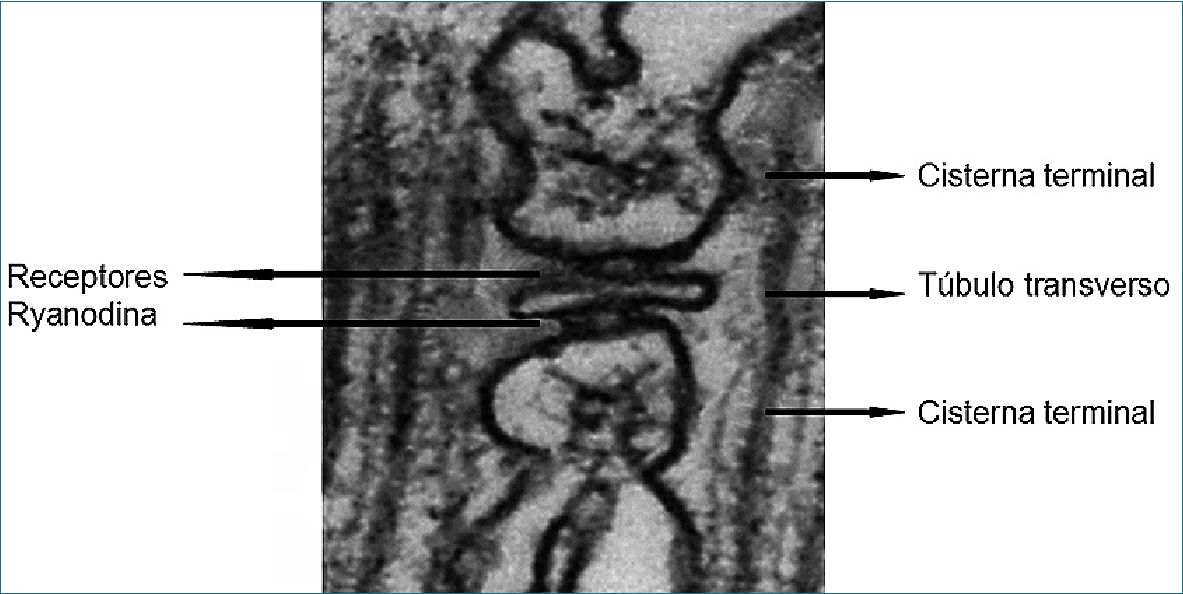
Figura 1. Elementos de la tríada: túbulo transversal y dos cisternas terminales adyacentes.
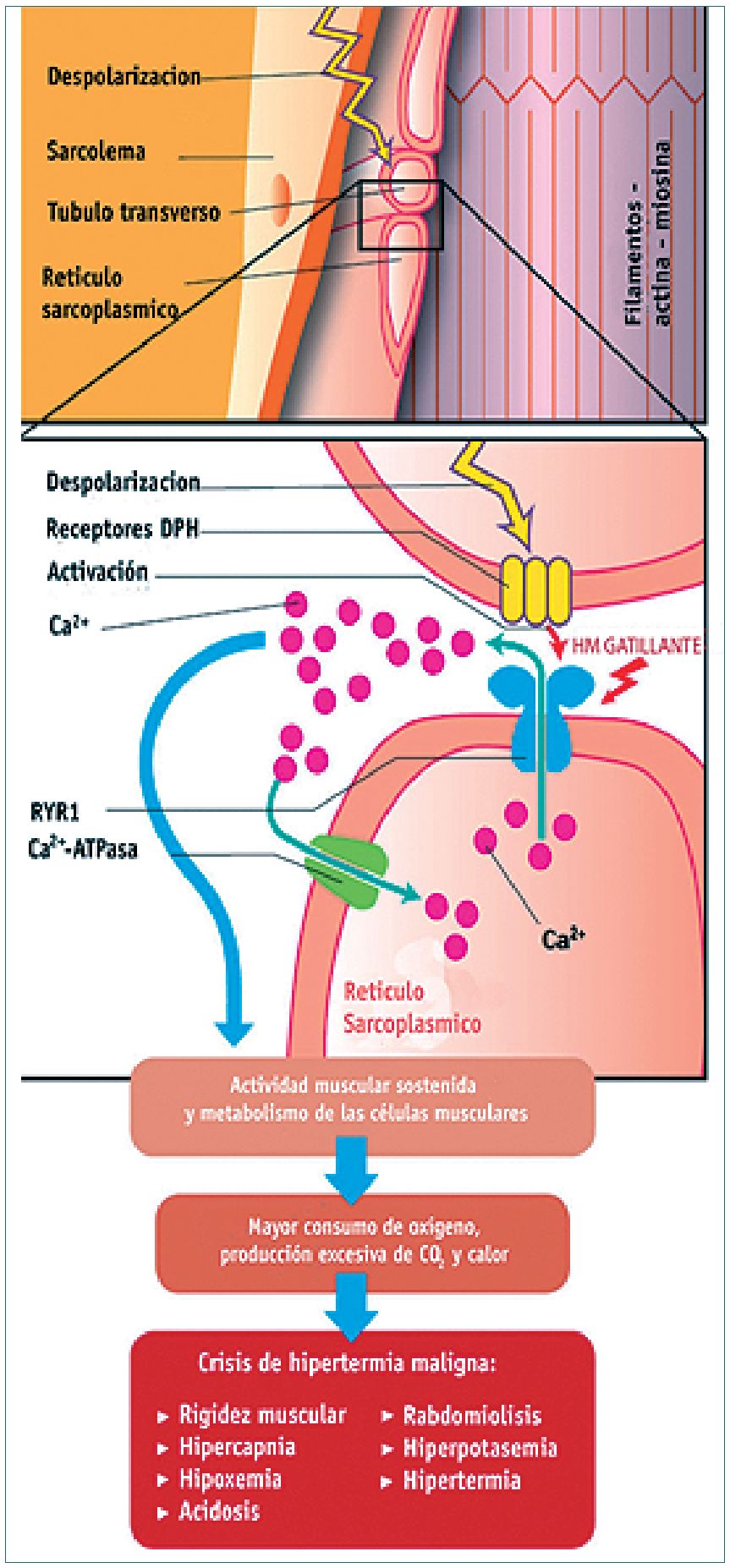
Figura 2. Cambios fisiopatológicos durante una crisis de hipertermia maligna.
En los mamíferos se conocen 3 isoformas de receptor de ryanodona (llamados así porque se unen al alcaloide ryanodina): RYR1, ubicada predominante en el músculo esquelético; RYR2, expresado en el músculo cardiaco y RYR3, que se encuentra el sistema nervioso central y en el músculo esquelético y liso[6].
Para hacerse una idea de la complejidad del tema, el gen RyR1, se ubica en el cromosoma 19 en el locus 19q13.1 (Figura 3). Se trata de un gen complejo ubicado en el brazo largo del cromosoma 19, que está compuesto por 106 exones (región de un gen que contiene la información para producir la proteína del gen; cada exón codifica una porción específica de la proteína completa) transcritos bajo la forma de ARN mensajero de 15.364 nucleótidos. Se trata de uno de los genes humanos más complicados, por lo que la mayoría de los laboratorios pueden observar mutaciones sólo en pequeñas porciones del gen.
Las mutaciones en el gen RYR1 en el cromosoma 19q13.1 se han asociado con una predisposición a HM. Se han identificado más de 300 variantes de la isoforma RYR1 y, por lo menos 31 de ellas, se han ensayado funcionalmente en estudios intracelulares de liberación de calcio, confirmándose responsables de HM[7]. Solo en el 50% de los pacientes susceptibles a HM, se detectó una mutación en RYR1[8].
Otras mutaciones importantes son las que ocurren en el gen, CACNA1S, que codifica la subunidad alfa del receptor de dihidropiridina dependientes de voltaje (DHPR), localizada en el cromosoma 1q32[9].
En resumen, la HM es un trastorno genético de gran heterogeneidad, en que se ha encontrado alteraciones en varios loci de diferentes cromosomas en diferentes familias, no siempre recurrentes y con gran dispersión. Esto concuerda con el hecho de que el control del calcio intracelular es un fenómeno complejo que no depende de la función de un solo organelo y hace muy difícil el diagnóstico en base al estudio genético.
-
Presentación clínica
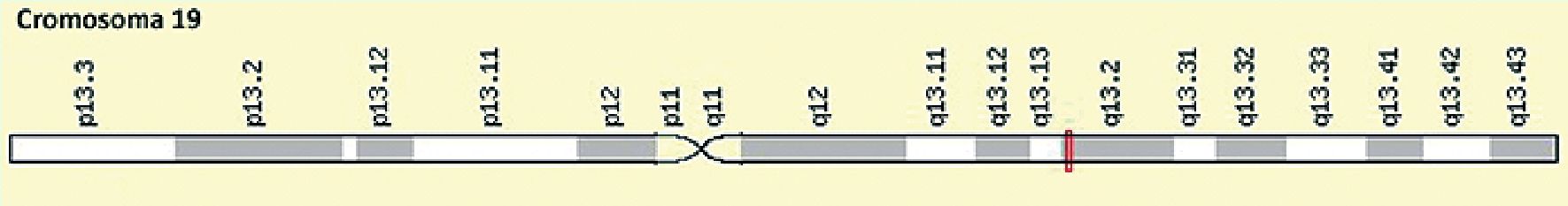
Figura 3. Mapa del cromosoma 19: Ubicación genómica del gen RyR1 (19q13.1).
La velocidad de aparición de la crisis de HM es diferente según el agente halogenado utilizado. Sin usar succinilcolina, la velocidad media de aparición con halotano es de 35 minutos, con isoflurano 140 minutos y con desflurano 260 minutos. Se ha reportado HM después de 7 horas de uso de desflurano. Probablemente debido a esta diferencia en el tiempo de aparición del cuadro con los diferentes agentes desencadenantes, la tendencia es que con los nuevos agentes inhalatorios los episodios ocurran más frecuentemente hacia el final de la anestesia o en la sala de recuperación. La administración de succinilcolina seguida de un anestésico inhalatorio es más probable que produzca espasmo de maséteros y el desarrollo de un cuadro de HM más rápido y severo.
Los síntomas clínicos de la HM son muy variables, desde leves o moderados hasta crisis fulminantes con hipermetabolismo muscular severo y rabdomiólisis. La capnografía y la oximetría tiene un gran valor diagnóstico.
La ETCO2 aumenta progresivamente, pudiendo llegar en unos pocos minutos a sobrepasar los 80 mmHg y los gases en sangre arterial revelan una acidosis respiratoria. Cifras de ETCO2 mayores de 55 mmHg o de PaCO2 mayores de 60 mmHg a pesar de una ventilación adecuada tienen un alto valor diagnóstico. Esta hipercarbia constituye un signo precoz, sensible y específico de HM. A raíz de la disminución del uso de succinilcolina se ha observado un aumento más gradual del ETCO2[10].
Aunque la ventilación mecánica permite mantener una adecuada saturación de oxígeno, el cirujano puede apreciar sangre venosa oscura en el campo operatorio, producto de una caída de la saturación venosa de oxígeno. El consumo de oxígeno aumenta 3 a 5 veces. La hipoxemia es un signo tardío en el curso de un episodio de HM.
Otros síntomas tempranos de una crisis de HM pueden incluir taquicardia sinusal, arritmia supraventricular o ventricular y espasmo de maséteros aislado o rigidez muscular generalizada. Una taquicardia sinusal inespecífica puede malinterpretarse como anestesia superficial, lo que a menudo retrasa el diagnóstico de HM, o viceversa, una anestesia superficial puede sobrediagnosticar una crisis de HM.
La elevación de la temperatura corporal es un signo tardío y dramático de HM, aunque en algunas series, han encontrado que la hipertermia estaba entre los primeros 3 signos más precoces en el 63,5% de las reacciones de HM[11].
La gasometría arterial revela una combinación de acidosis metabólica y respiratoria con exceso de base negativo, aumento del lactato, hipercapnia e hipoxemia. La crisis fulminante de HM generalmente se presenta con valores de paCO2 > 60 mmHg y un deficit de base > 8.
A medida que avanza el episodio de HM, la rabdomiólisis conduce a hiperpotasemia, aumento de la creatin fosfoquinasa y mioglobinemia, que puede provocar insuficiencia renal aguda. Las complicaciones adicionales potencialmente mortales incluyen la coagulación intravascular diseminada, insuficiencia cardiaca congestiva, isquemia intestinal y síndrome compartimental de extremidades. La etapa final de una crisis de HM fulminante se caracteriza por un fallo multiorgánico y un colapso circulatorio.
-
Contractura de maséteros: estado actual
El síndrome muscular se manifiesta en forma de rigidez muscular y rabdomiólisis. La rigidez muscular puede ser precoz, a nivel de los maséteros o de la musculatura de los miembros inferiores, o puede ser más tardía y generalizada, dificultando incluso la ventilación mecánica.
En algunos pacientes, especialmente en niños, la succinilcolina puede producir un cuadro de contractura de los músculos maséteros durante la inducción de la anestesia, más allá de la contracción habitual de los músculos de la mandíbula con las fasciculaciones, que hace la intubación difícil o imposible. En niños inducidos con halotano y succinilcolina tiene una incidencia de 0,3% a 1%. La base de datos de la MHAUS (del inglés, Malignant Hyperthermia Association of the United States) reporta que los hombres tienen 2,8 veces más probabilidad de desarrollar hipertermia maligna por contractura de los maséteros y los pacientes sometidos a cirugía ortopédica 3,3 veces más que los sometidos a otro tipo de cirugía[12].
La definición de rigidez de los maséteros es clínica y subjetiva, pues se ha reportado un aumento transitorio y moderado del tono de los músculos maséteros como un efecto “normal” de la succinilcolina en los niños y, se ha descrito una relajación incompleta hasta en el 4,4% de los casos. Este signo clínico ha sido considerado como precursor de HM, pues su incidencia es mucho mayor en este grupo que en la población general, especialmente cuando es intenso y prolongado. Los pacientes con contractura de maséteros y rigidez muscular generalizada después de la administración de succinilcolina tienen mayor probabilidad de ser HMS que los que tienen una moderada y aislada contractura de maséteros.
Se ha planteado dividir el espasmo de los maséteros secundario a la administración de succinilcolina en tres formas clínicas de presentación, que se complementan con tres opciones de manejo del cuadro: Si las mandíbulas logran abrirse completamente mediante la presión manual y el paciente puede ser intubado sin dificultad, pueden usarse agentes desencadenantes y monitorizar al paciente en forma rutinaria.
– Si las mandíbulas se abren solo parcialmente haciendo difícil la intubación, debe sospecharse el cuadro de HM, utilizando sólo agentes no desencadenantes y monitorizando la eventual aparición de una crisis.
– Si la boca no puede abrirse, puede haber comenzado una crisis de HM y se sugiere detener inmediatamente la anestesia y postergar la cirugía.
La urgencia de la cirugía juega un papel importante en la toma de decisiones: si se trata de una operación electiva es recomendable la suspensión de la cirugía y el estudio de susceptibilidad a hipertermia maligna; si se trata de una cirugía de urgencia debe continuarse la anestesia con agentes seguros (no desencadenantes) y debe vigilarse estrechamente la aparición de otros signos. Si el anestesiólogo y el cirujano están de acuerdo en proseguir con la cirugía, debe hacerse con el sobreentendido de que puede ser necesario acortarla o interrumpirla si se detecta alguna manifestación clínica de HM.
En los últimos años la incidencia de este cuadro ha bajado, probablemente debido a la disminución del uso de succinilcolina en los niños, por las recomendaciones del etiquetado de la droga avisando su potencial peligro.
-
Escala de clasificación clínica
Larach, desarrolló una escala de clasificación clínica para ayudar al diagnóstico clínico del síndrome en base a un sistema de puntuación. Se da una ponderación diferencial a cada una de las manifestaciones clínicas de la enfermedad (Tabla 1)[13].
Basta con la presencia de un solo elemento en cada proceso para calificar para la puntuación. A partir de ese momento, se genera un puntaje que evalúa la probabilidad de que el episodio se trate de una HM en una escala de “casi nunca a casi seguro”. Tratándose ésta de una escala clínica y dependiendo del resultado de pruebas de laboratorio, su valor reside en identificar a aquellos sujetos con los episodios más convincentes de HM para una posterior evaluación con pruebas diagnósticas. En realidad, es un verdadero ranking de susceptibilidad a HM.
Hasta ahora, sin embargo, la escala no ha mostrado tener un gran acierto. Un estudio alemán demostró que, de 92 pacientes, sólo 32 (35%) fueron diagnosticados como susceptibles a HM con la prueba de contractura in vitro (IVCT, del inglés in vitro contracture test), 47 (51%) no tenían HM y 13 (14%) eran equívocos a HM[14].
-
Diagnóstico
El diagnóstico de la crisis es clínico, en lo posible antes de la aparición de hipertermia; la capnografía y la oximetría han permitido hacerlo más precozmente. Después de la crisis o en los pacientes posiblemente susceptibles, el diagnóstico es de laboratorio.
El gold standard para el diagnóstico de laboratorio de HM es el IVCT, basado en la contracción de fibras musculares tomadas a partir de una biopsia de músculo estriado en presencia de halotano o cafeína. Dos normas son ampliamente usadas: una por el grupo de HM europeo (EMMG, del inglés European Malignant Hyperpyrexia Group) y la otra por el grupo de HM estadounidense (NAMHR, del inglés North American Malignant Hyperthermia Registry)[15],[16].
Usando el protocolo europeo, una persona se considera MHS cuando los resultados de las pruebas de cafeína y halotano son positivos. Se obtiene un diagnóstico de MH normal (MHN) cuando ambas pruebas son negativas. Un tercer diagnóstico, MH equívoco (MHE), se obtiene cuando solo uno de los análisis (halotano o cafeína) es positivo.
Usando el protocolo estadounidense, a una persona se le diagnostica MHS cuando cualquiera de las pruebas de halotano o cafeína es positiva, y MHN cuando ambas pruebas son negativas.
El protocolo EMHG puede reducir la posibilidad de resultados falsos positivos y negativos en comparación con el protocolo NAMHR, pero en general se obtienen resultados similares. El protocolo EMHG tiene una sensibilidad del 99% y una especificidad del 94%, mientras que las cifras de sensibilidad y especificidad para el NAMHR son de 97% y 78% respectivamente.
La IVCT es costosa, se limita a los centros de pruebas especializados, requiere un procedimiento quirúrgico y puede producir resultados equívocos, falsos positivos y negativos.
El análisis de ADN, que requiere sólo una muestra de sangre, ofrece una alternativa al IVCT, sin embargo, no puede reemplazar a las pruebas de contractura muscular in vitro, debido a la heterogeneidad de la HM con relación al número de variantes del gen RYR1 y al involucramiento de otros genes. Adicionalmente, el diagnóstico basado en ADN para susceptibilidad a HM puede ser llevado a cabo solamente en familias con mutaciones con características funcionales. Un diagnóstico negativo para HM no puede ser hecho exclusivamente con una prueba de ADN[17].
| Tabla 1. Indicadores clínicos para determinación de hipertermia maligna | |
| Proceso | Indicador |
| I. Rigidez | a. Rigidez muscular generalizada
b. Espasmo de maséteros tras administración de succinilcolina |
| II. Destrucción muscular | a. CK > 20.000 UI»l-1 después de anestesia con succinilcolina b. CK > 10.000 UI»l-1 después de anestesia sin succinilcolina c. Coluria en el postoperatorio
d. Mioglobinuria > 60 ug»l-1. e. Mioglobinemia > 170 ug^l- f. Kalemia > 6 mEq^l- |
| III. Acidosis respiratoria | a. ETCO2 > 55 mmHg con ventilación controlada adecuada b. PaCO2 > 60 mmHg con ventilación controlada adecuada c. ETCO2 >60 mmHg en ventilación espontánea
d. PaCO2 >65 mmHg en ventilación espontánea e. Hipercarbia inapropiada f. Taquipnea inapropiada |
| IV. Aumento temperatura | a. Incremento inadecuado de la temperatura
b. Aumento inadecuado de la temperatura > 38,8 °C en el periodo perioperatorio |
| V. Afectación cardiaca | a. Taquicardia sinusal inadecuada
b. Taquicardia o fibrilación ventriculares |
| CK: Creatina fosfoquinasa o CPK. | |
Las indicaciones para realizar una prueba de contractura son:
– Pacientes con historia personal sospechosa de HM.
– Parientes de primer grado de un paciente con historial sospechoso.
– Pacientes portadores de alguna miopatía predisponente de HM.
Las miopatías relacionadas con HM han ido variando a lo largo del tiempo, de acuerdo con la evidencia. Actualmente, se incluye en la lista de aquellas que están definitivamente relacionadas: la enfermedad del núcleo central, la enfermedad del multiminicore y el síndrome o fenotipo de King-Denborough. Otras enfermedades producen un síndrome similar a HM: distrofias musculares o distrofiopatías (enfermedades de Becker y Duchenne), enfermedades de canales iónicos y rabdomiólisis inducida por ejercicio o calor. Finalmente, hay enfermedades que actualmente tienen una evidencia muy leve o no tienen evidencia de estar relacionadas con HM: síndrome de Noonan, osteogénesis imperfecta, artrogriposis y miopatías mitocondriales[18],[19].
En la Tabla 2 se adjunta un listado de los centros en los que se pueden realizar las biopsias diagnósticas para los tests de contractura en Estados Unidos de Norteamérica. Los centros más cercanos, sin embargo, están en Brasil. Tanto el Centro de Biopsia Muscular de la Universidad Federal de Río de Janeiro como el Cedhima (Centro de Estudio, Diagnóstico e Investigación de Hipertermia Maligna) de la Facultad de Medicina de la Universidad de Sao Paulo, usan con mucho éxito el protocolo norteamericano (NAMHR) para el diagnóstico de HM[20].
-
Prevención de la crisis
Los pacientes que han sido diagnosticados como MHS, o más comúnmente en Chile, aquellos que han tenido una crisis de HM previa o tienen parientes consanguíneos con antecedentes de HM o muerte en pabellón de causa sospechosa, deben ser tratados en forma especialmente cuidadosa. Muchos de estos pacientes han sido sometidos sin incidentes a intervenciones como tratamientos dentales con anestesia local y anestesia obstétrica antes de que se hiciese el diagnóstico de susceptibilidad.
Se debe preferir las técnicas neuroaxiales y regionales si la cirugía lo permite. En caso contrario debe disponerse de métodos anestésicos libres de agentes desencadenantes, como infusores de anestesia endovenosa total. La evidencia actual es que no está recomendada la profilaxis con dantroleno.
El pabellón debe ser preparado y el personal debe ser adiestrado con el objetivo de evitar la administración inadvertida o accidental de agentes desencadenantes. Para ello, se recomienda destacar que ese pabellón será para un paciente susceptible a HM (por ejemplo, colocar un letrero en puerta del pabellón). Deben ser retirados de la máquina de anestesia los vaporizadores de gases anestésicos y de los estantes de drogas las ampollas de succinilcolina[21]. Debe cambiarse el absorbente de CO2 (cal sodada) y verificar que los circuitos desechables estén nuevos.
La máquina de anestesia debe ser limpiada con un flujo alto de oxígeno durante 10 minutos a 2 horas según las especificaciones de cada dispositivo. Este proceso de preparación también debe hacerse en función de los agentes volátiles halogenados que se hayan utilizado en las últimas intervenciones. Jones et al., demostraron que el desflurano requería un tiempo de purga más largo que el sevoflurano, tanto en la máquina Datex-Ohmeda Aestiva como en la Aisys[22]. Se ha demostrado que la aplicación de filtros de carbón activado acelera un proceso de limpieza exitoso. Tales filtros deberían colocarse tanto en el extremo inspiratorio como en el extremo espiratorio de la máquina de anestesia y reemplazarse con un nuevo juego cada 60 min. La MHAUS, recomienda limpiar y preparar la estación de trabajo de anestesia de acuerdo con las recomendaciones del fabricante o los estudios publicados. Durante la administración de los gases, el hecho de bajar el flujo de gas fresco tras el período de lavado, puede permitir que la concentración del anestésico volátil se reacumule. El flujo de gas fresco debería mantenerse al menos a 10 l·min-1para evitar este rebote.
Para que el anestesiólogo enfrente con confianza la anestesia en los pacientes susceptibles, debe tener el conocimiento y contar con las habilidades para manejar una crisis de HM.
Los procedimientos ambulatorios son factibles en la mayor parte de los centros y el momento de alta depende de los criterios utilizados en cada centro uno. Sin embargo, cuando se administre anestesia general utilizando alguno de los agentes desencadenantes, tanto en el medio ambulatorio como en el hospitalario, debe haber dantrolene inmediatamente disponible. La recomendación actual es contar con un stock de 36 frascos, dosis necesaria para tratar a dosis máximas, una crisis de HM en un paciente de 70 kg de peso.
-
Tratamiento
Existen tres medidas principales en el manejo de la crisis de HM que constituyen una urgencia terapéutica, de modo que el resultado del tratamiento va a depender directamente de la velocidad de su puesta en marcha: 1) la detención de la administración de halogenados; 2) la hiperventilación con oxígeno al 100% y 3) la administración de dantrolene endovenoso.
Una vez sospechada la crisis con las manifestaciones clínicas de HM, descartando los posibles diagnósticos diferenciales (frecuentemente la superficialidad anestésica), se debe pedir ayuda y discontinuar los agentes desencadenantes. La detención de la administración de halogenados incluye la extracción de los vaporizadores y la continuación de la anestesia con agentes no desencadenantes. En acuerdo con el cirujano y conforme a las condiciones del paciente, debe decidirse si se suspende o no la cirugía en caso de que esto sea posible.
Con la finalidad de eliminar los gases circulantes, se recomienda hiperventilar, con O2 al 100% y con un flujo libre de gas de al menos 10 l·min-1. Aunque se trata de una de las medidas más importantes, es la más frecuentemente olvidada en todos los estudios de simulación. No es imprescindible cambiar el ventilador de la máquina de anestesia ni la máquina de anestesia completa. Tampoco ha demostrado ser beneficiosa la ventilación manual. Conjuntamente debe iniciarse de manera rápida la preparación y administración de dantrolene, el tratamiento específico de la crisis de HM.
| Tabla 2. Centros en Estados Unidos especializados en el diagnóstico de susceptibilidad a Hlm. Los centros más cercanos en Brasil están especificados en el texto |
| Toronto General Hospital |
| Malignant Hyperthermia Investigation Unit Eaton 3-323 200 Elizabeth St.
Toronto, Ontario M5G 2C4 PH: (416) 340-3128 Email: sheila.riazi@uhn.ca |
| Uniformed Services University of the Health Sciences |
| Bethesda, MD (Military & Civilian)
LCDR Michael Lee MC, USN CAPT Dale F. Szpisjak MC, USN (back up) Department of Anesthesiology PH: (301) 295-3140 Email: MHLab@usuhs.edu |
| UC Davis mH Biopsy Testing Center |
| Sacramento, CA
Timothy Tautz, MD PH: (916) 734-2431 Email: tjtautz@ucdavis.edu |
| University of minnesota |
| Minneapolis, MN
Paul A. laizzo, PhD PH: (612) 624-7912 or -3959 Email: iaizz001@umn.edu |
| Wake Forest Baptist medical Center |
| Winston-Salem, NC
Sherry Meacham PH: (336) 716-7194 Email: smeacham@wakehealth.edu |
El dantrolene es un derivado de la hidantoína (1-[[[5-(4-nitrofenilo)-2-furanilo]metileno]imino]-2,4imidazolidinediona) que, aunque no es un bloqueador neuromuscular, produce debilidad muscular, por acción directa sobre el músculo estriado. Esta potenciación puede hacer necesaria la ventilación mecánica en el postoperatorio si no se ha logrado un completo antagonismo del bloqueo neuromuscular. Su acción se ha relacionado con la reducción del flujo de salida de Ca++ desde el retículo sarcoplásmico, contrarrestando la disminución de la inhibición del músculo esquelético producida por Mg2+ en el músculo afectado por HM. El dantrolene puede inhibir la sensibilidad aumentada a la cafeína observada en músculos con HM, y se ha demostrado que disminuye la liberación de Ca++ inducida por despolarización.
Antes de su administración, el dantrolene, originalmente liofilizado, debe ser reconstituido en agua bidestilada o manitol (en solución salina precipita) y suministrado rápidamente al paciente. La reconstitución del fármaco es más sencilla a mayor temperatura lo que se transforma en un hecho importante ya que el tiempo de preparación es un factor vital.
Un frasco de dantroleno de 20 mg debe diluirse en 60 ml de agua bidestilada o manitol. La dosis inicial es de 2,5 mg·kg-1 de dantrolene en lo posible a través de una vía central (la falta de una línea central no debe retrasar el tratamiento). La dosis debe repetirse cada 10 minutos las veces necesarias hasta la remisión total de los síntomas. Puede llegar a administrarse 10 mg·kg , o más, por eso la necesidad de un stock de 36 frascos.
Paralelamente al tratamiento etiológico, se debe corregir las alteraciones fisiopatológicas producidas por la HM.
La acidosis metabólica debe ser corregida administrando 1-4 miEq^kg de bicarbonato de sodio 2/3 molar IV, con metas guiadas por gasometría. A su vez, esto contribuye a producir alcalinización de la orina y, con esto, protección del daño renal inducido por la mioglobinuria. Se recomienda mantener una diuresis mayor de 1 ml·kg-1·h-1, lo que implica la necesidad de instalar una sonda de Foley, para un adecuado control de este objetivo.
La temperatura también es un objetivo del manejo. Para esto se utilizan medidas como la administración de soluciones IV frías y el enfriamiento de la superficie y las cavidades corporales con fluidos helados. La temperatura debe monitorizarse estrictamente con termómetro central, con el objetivo de no producir una hipotermia inadvertida.
Las arritmias por lo general se previenen o tratan con el manejo de la acidosis e hiperkalemia, pero en caso de persistir deben manejarse según protocolo ACLS, evitando los bloqueadores de calcio ya que pueden aumentar los niveles de potasio sanguíneo. Si la hiperkalemia persiste en rangos que pongan en riesgo la vida del paciente, se deben usar medidas clásicas del manejo de ésta, que incluyen el uso de calcio, bicarbonato, insulina y glucosa (Tabla 3).
Durante la crisis y en el postoperatorio inmediato deben controlarse periódicamente algunos exámenes de laboratorio: gases en sangre, electrolitos plasmáticos, creatininemia, lactacidemia, mioglobinemia, estudio de coagulación, pruebas hepáticas y CK.
En la página web de la Sociedad de Anestesiología de Chile existe un excelente resumen sobre el “Manejo de la Crisis de Hipertermia Maligna y para el Manejo del Paciente Susceptible de Hipertermia Maligna”, que sería recomendable tener en todas las máquinas de anestesia. Un buen recordatorio en un momento de crisis se agradece, especialmente en situaciones de tan baja prevalencia[23].
La recidiva de HM es de aproximadamente 50%, normalmente en las primeras 6,5 horas después de la crisis, por lo que estos pacientes deben ser observados en una unidad de cuidados intensivos por lo menos durante 24 h. Por esta razón, se recomienda mantener una infusión de dantroleno de 0,25 mg·kg-1·h-1 durante las primeras 6 h. Después se puede administrar 1 mg·kg-1 de dantroleno por vía oral cada 6 horas, las primeras 24 horas.
Debe tenerse precaución con la administración de ondansetrón en el intra y postoperatorio. Como antagonista de la serotonina, el ondansetrón puede aumentar la serotonina en el receptor 5-HT2A en el espacio presináptico. En un individuo susceptible, el agonismo del receptor 5-HT2A puede precipitar una crisis de HM[24].
-
Conclusiones
La evaluación preoperatoria al lado del paciente es una herramienta importantísima para la detección de pacientes susceptibles, aunque deja fuera un gran porcentaje de pacientes sin historia o que han recibido anestesias previamente sin presentar problemas.
| Tabla 3. Esquema de manejo de hiperkalemia para niveles de K sanguíneo > 5,9 mEqd-1 o menores si producen arritmias | |
| Agente
Cloruro de calcio |
Tratamiento
10 mg·kg-1 (máximo 2 g) |
| Gluconato de calcio | 30 mg·k-1 (máximo 3 g) |
| Bicarbonato de sodio | 1-4 mEq·k-1 (máximo 50 mEq) |
| Glucosa + insulina | Adultos: 10 u insulina con 50 ml de suero glucosado 50% IV
– Niños: 0,1u insulina con 0,5 g·k-1 de glucosa – Control de glicemia horaria |
Lamentablemente, en nuestra realidad, hacer estudios diagnósticos es poco probable. Además, debido a su compleja forma de herencia, la mayoría de las veces será una situación sorpresiva, frente a la cual todo anestesiólogo debe estar preparado. Esto hace destacar la importancia de la constante capacitación en centros de simulación y la revisión permanente de medicamentos para manejo de la crisis, así como la existencia de protocolos de prevención cuando se sospeche.
En la actualidad, con un adecuado diagnóstico y manejo eficaz, la sobrevida es muy alta, pese a ser un evento mortal si se deja evolucionar espontáneamente. Anteriormente se estimaba que la mortalidad se elevaba al 80%. Actualmente ha descendido a niveles cercanos al 5%, gracias al uso del dantrolene y al progreso de la monitorización durante el acto anestésico, lo que permite que el diagnóstico sea precoz[25].
Referencias
1. Bustamante R, Covarrubias MS. Hipertermia Maligna. Revista de la Asistencia Pública de Santiago. 1982;1:14–23.
2. Brady JE, Sun LS, Rosenberg H, Li G. Prevalence of malignant hyperthermia due to anesthesia in New York State, 2001-2005. Anesth Analg. 2009 Oct;109(4):1162–6. https://doi.org/10.1213/ane.0b013e3181ac1548 PMID:19762744
3. Larach MG, Gronert GA, Allen GC, Brandom BW, Lehman EB. Clinical presentation, treatment, and complications of malignant hyperthermia in North America from 1987 to 2006. Anesth Analg. 2010 Feb;110(2):498–507. https://doi.org/10.1213/ANE.0b013e3181c6b9b2 PMID:20081135
4. Struk A, Lehmann-Horn F, Melzer W. Voltage-dependent calcium release in human malignant hyperthermia muscle fibers. Biophys J. 1998; 75:2 402–10. https://doi.org/10.1016/S0006-3495(98)77684-2.
5. Schneiderbanger D, Johannsen S, Roewer N, Schuster F. Management of malignant hyperthermia: diagnosis and treatment. Ther Clin Risk Manag. 2014 May;10:355–62. PMID:24868161
6. Hamilton SL. Ryanodine receptors. Cell Calcium. 2005 Sep-Oct;38(3-4):253–60. https://doi.org/10.1016/j.ceca.2005.06.037 PMID:16115682
7. European Malignant Hyperthermia Group. Diagnostic MH Mutations. Disponible en: https://www.emhg.org/diagnostic-mutations
8. Rosenberg H. Mining for mutations in malignant hyperthermia. Anesth Analg. 2011 Nov;113(5):975–6. https://doi.org/10.1213/ANE.0b013e31822dd4ca PMID:22021794
9. Carpenter D, Ringrose C, Leo V, Morris A, Robinson RL, Halsall PJ, et al. The role of CACNA1S in predisposition to malignant hyperthermia. BMC Med Genet. 2009 Oct;10(1):104. https://doi.org/10.1186/1471-2350-10-104 PMID:19825159
10. Pollock AN, Langton EE, Couchman K, Stowell KM, Waddington M. Suspected malignant hyperthermia reactions in New Zealand. Anaesth Intensive Care. 2002 Aug;30(4):453–61. https://doi.org/10.1177/0310057X0203000410 PMID:12180584
11. Larach MG, Allen GC, Brandom BW. Temperature changes are not late signs of malignant hyphertermia: a NAMH registry of MHAUS study. Anesthesiology. 2008;109:318–38.
12. Base de datos de la MHAUS, en https://www.mhaus.org/cfw/
13. Larach MG, Localio AR, Allen GC, Denborough MA, Ellis FR, Gronert GA, et al. A clinical grading scale to predict malignant hyperthermia susceptibility. Anesthesiology. 1994 Apr;80(4):771–9. https://doi.org/10.1097/00000542-199404000-00008 PMID:8024130
14. von Richthofen V, Wappler F, Scholz J, Fiege M, Schulte am Esch J. [Evaluation of malignant hyperthermia episodes with the Clinical Grading Scale]. Anasthesiol Intensivmed Notfallmed Schmerzther. 1998 Apr;33(4):244–9. https://doi.org/10.1055/s-2007-994239 PMID:9617423
15. The European Malignant Hyperpyrexia Group. A protocol for the investigation of malignant hyperpyrexia (MH) susceptibility. Br J Anaesth. 1984 Nov;56(11):1267–9. https://doi.org/10.1093/bja/56.11.1267 PMID:6487446
16. Larach MG, Landis JR, Bunn JS, Diaz M. Prediction of malignant hyperthermia susceptibility in low-risk subjects. An epidemiologic investigation of caffeine halothane contracture responses. The North American Malignant Hyperthermia Registry. Anesthesiology. 1992 Jan;76(1):16–27. https://doi.org/10.1097/00000542-199201000-00003 PMID:1729931
17. Hopkins PM, Rüffert H, Snoeck MM, Girard T, Glahn KP, Ellis FR, et al.; European Malignant Hyperthermia Group. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth. 2015 Oct;115(4):531–9. https://doi.org/10.1093/bja/aev225 PMID:26188342
18. Litman RS, Griggs SM, Dowling JJ, Riazi S. Malignant Hyperthermia Susceptibility and Related Diseases. Anesthesiology. 2018 Jan;128(1):159–67. https://doi.org/10.1097/ALN.0000000000001877 PMID:28902673
19. Corvetto M, Heider R, Cavallieri S. Hipertermia maligna: ¿cómo estar preparados? Rev Chil Cir 2013; 65 versión On-line ISSN 0718-4026
20. de Carvalho Correia AC, Barros Silva PC, Araújo da Silva B. Hipertermia Maligna: Aspectos Moleculares y Clínicos. Rev Bras Anestesiol. 2012;62(6):1–10.
21. Malignant Hyperthermia Association of the United States. How to be prepared. Disponible en: https://www.mhaus.org/healthcare-professionals/be-prepared/
22. Jones C, Bennett K, Kim TW, Bulger TF, Pollock N. Preparation of Datex-Ohmeda Aestiva and Aisys anaesthetic machines for use in malignant hyperthermia susceptible patients. Anaesth Intensive Care. 2012 May;40(3):490–7. https://doi.org/10.1177/0310057X1204000315 PMID:22577915
23. Sociedad de Anestesiología de Chile. Disponible en https://www.sachile.cl/view/pdf_pages/manejo_de_la_crisis_de_hipertermia_maligna.pdf/
24. Johannsen S, Roewer N, Schuster F. Ondansetron-induced muscular contractures in malignant hyperthermia-susceptible individuals. Anesth Analg. 2012 Oct;115(4):925–8. https://doi.org/10.1213/ANE.0b013e31825d3749 PMID:22696611
25. Glahn KP, Ellis FR, Halsall PJ, Müller CR, Snoeck MM, Urwyler A, et al.; European Malignant Hyperthermia Group. Recognizing and managing a malignant hyperthermia crisis: guidelines from the European Malignant Hyperthermia Group. Br J Anaesth. 2010 Oct;105(4):417–20. https://doi.org/10.1093/bja/aeq243 PMID:20837722