Aurio Fajardo1,a, Alberto Medina2,a, Vicent Modesto3,a, Federico Gordo-Vidal1 2 3 4
 ORCID
ORCIDRecibido: 02-10-2020
Aceptado: 09-10-2020
©2020 El(los) Autor(es) – Esta publicación es Órgano oficial de la Sociedad de Anestesiología de Chile
Revista Chilena de Anestesia Vol. 49 Núm. 6 pp. 784-794|https://doi.org/10.25237/revchilanestv49n06-04
PDF|ePub|RIS
The dark side of hypoxemia
Abstract
Acute respiratory distress syndrome (ARDS) is characterized by an increase in the intrapulmonary shunt (measured by the PaO2/FiO2 ratio) caused by bilateral alveolar-interstitial infiltrates which are not fully explained by fluid overload. However, there are some ARDS cases which present severe hypoxemia without clear lung infiltrates. An example of this, which has generated great controver- sy, is the infection caused by SARS-CoV-2. Understanding the pathophysiology of hypoxemia is absolutely crucial in order to establish the most appropriate therapeutic strategy for each patient. In the case of a severe hypoxemia (PaO/ FiO2 < 200 which represents a shunt greater than 30%) with a chest X-ray (or CT) without clear and extensive bilateral infiltrates, it is important to consider that the shunt can be caused due to vascular involvement. This shunt could be explained two ways: an increase in pulmonary vascular resistance (PVR), which generates a right-to-left shunt through the patent foramen ovale (PFO), or an alteration of the hypoxic pulmonary vasoconstriction reflex (HPV). The HPV reflex is activated in an attempt to redistribute the vascular flow to better ventilated areas. However, there are some situations (such as viral infections) that can alter this reflex and worsen the hypoxemia. The concomitant use of vasoactive drugs (such as inhaled nitric oxide) and vasopressors (such as dopa- mine or norepinephrine) has been proposed with the aim of reducing PVR and the flow through the PFO; or to redistribute the flow to better ventilated areas if an alteration of the RVP is suspected.
Resumen
Un síndrome de distrés respiratorio agudo (SDRA) se caracteriza por un incremento del intrapulmonar (medido por el cociente PaO2/FiO2) causado por una afectación alveolo-intersticial bilateral no explicada por sobrecarga hídrica. Sin embargo, hay casos de SDRA que presentan una marcada hipoxemia sin claros infiltrados pulmonares. Un ejemplo de este caso, que ha generado gran controversia, es la infección por SARS-CoV-2. El entendimiento de la fisiopato- logía de la hipoxemia es absolutamente clave para establecer la estrategia terapéutica más adecuada en cada paciente. Ante una hipoxemia grave (PaO2/FiO2 < 200 que representa un superior al 30%) y con una radiografía de tórax (o con TAC) sin claros y extensos infiltrados bilaterales, el podría deberse a una afectación vascular. Esto podría explicarse por dos causas: un aumento de las resistencias vasculares pulmonares (RVP), que genera un derecha- izquierda a través del foramen oval permeable (FOP) o una alteración del reflejo de vasoconstricción pulmonar hipóxico (VPH). El reflejo de VPH se activa en un intento por redistribuir el flujo vascular hacia las zonas mejor ventiladas. Sin embargo, existen situaciones (como infecciones víricas) que pueden alterar dicho reflejo y agravar la hipoxemia. Se ha propuesto el uso concomitante de fármacos vasoactivos (como el óxido nítrico inhalado) y vasopresores (como do- pamina o noradrenalina) con el objetivo de disminuir las RVP y el flujo a través del FOP; o para redistribuir el flujo a zonas mejor ventiladas si se sospecha una alteración del RVP.
-
Introducción
Hermes Trismegisto, en su sexta ley, afirma que todo tiene una causalidad, mas no una casualidad. Toda causa tiene su efecto y todo efecto tiene su causa, y la magnitud del efecto es equivalente a la importancia de la causa que lo generó.
Ante una hipoxia, la microcirculación pulmonar experimenta un fenómeno adaptativo y exclusivo de vasoconstricción con el objetivo de redistribuir el flujo sanguíneo desde zonas pobremente aireadas hacia las que tengan mayor aireación, disminuyendo así el shunt intrapulmonar. La finalidad de este efecto es la optimización de la oxigenación.
Este fenómeno agudo de vasoconstricción pulmonar hipóxico (VPH) fue descrito hace más de un siglo y aún es motivo de masivas investigaciones fisiológicas. Los estudios iniciales se desarrollaron en modelos de circulación pulmonar felina[1 ] y en 1922 Haldane sugirió que el pulmón tiene la capacidad de autorregular su flujo vascular mediante contracción o dilatación de sus capilares[2].
En la insuficiencia respiratoria aguda por corona virus (SARS-CoV-2) algunos autores han tratado de explicar las fases evolutivas de esta patología mediante denominaciones de propia autoría[3 ], las cuales
han sido motivo de grandes debates.
Gattinoni et al.[4 ] sugirieron 2 fenotipos: L (low) y H (high) basándose en la elastancia y reclutabilidad pulmonar. Afirman que en el fenotipo L existe una alteración de la regulación de la perfusión por pérdida de la VPH. Sin embargo, en otra publicación, el mismo autor señala que habría una abolición del reflejo de VPH, incremento de la mezcla venosa y una “excesiva vasoconstricción pulmonar”[5 ].
Brochard et al.[6 ] describieron un síndrome de dis trés respiratorio agudo (SDRA) de presentación “atípica” y la probable existencia de un “subfenotipo” con alteración vascular (por pérdida de la vasoconstricción pulmonar) como mecanismo etiológico de la hipoxemia.
Lo cierto es que en esta pandemia la evidencia se está creando con la experiencia del día a día. A esta enfermedad se podría clasificar en estadios, aunque no sean necesariamente evolutivos:
1 .- “Estadio inicial”. El paciente presenta un patrón radiolúcido o hipodenso (radiografía y TAC respectivamente) en el que aún no existe (o existe, pero en mínima cantidad) el patrón de vidrio esmerilado periférico y bilateral característico de las neumopatías virales. En esta etapa aún no se hará evidente la insuficiencia respiratoria.
2 .- “Estadio tardío” (o SDRA clásico). La etapa clínicamente más florida, se explica por la exponencial e incontrolada respuesta inflamatoria, la activación de factores proinflamatorios con invasión alveolar y producción de exudado, edema y consolidaciones, activación de la cascada de coagulación, etc.
La progresión de la enfermedad se manifiesta tanto clínica como gasométricamente en forma distinta y el soporte dependerá del estadio. En relación con el soporte ventilatorio, no será lo mismo apoyar al paciente en estadio inicial que al paciente en estadio tardío.
Sin embargo, para saber el cómo y el por qué se debe brindar soporte a los pacientes en “estadio inicial” es imprescindible entender la fisiopatología pulmonar de esta etapa.
La alteración del reflejo de VPH es fundamental para entender la fisiopatología de la injuria pulmonar aguda. En la hipoxia por SARS-CoV-2 también ha sido descrita dicha alteración[7 ].
-
Sustrato fisiopatológico
West[8 ] considera que ante una hipoxia los capilares arteriales pulmonares experimentan una contracción de su musculatura lisa. Esta hipoxia está determinada por la presión arterial de oxígeno (PaO2) y por la oxigenación de la sangre venosa mixta (PvO2) que representa aproximadamente una quinta parte de la respuesta. Se describen 2 fases[9 ]:
1. Fase 1. Ante una hipoxia el reflejo se desencadena en cuestión de segundos, prolongándose por un máximo de 15 min.
a. Con una hipoxia moderada (PaO2 30-50 mmHg) la respuesta se vuelve sostenida.
b. Con una hipoxia grave (< 30 mmHg en estudios experimentales) la fase 1 termina rápidamente con niveles de oxemia casi normales.
2. Fase 2. Se inicia cuando la hipoxia moderada persiste por más de 30-60 min, desencadenando un incremento de la resistencia vascular pulmonar (RVP). Alcanza el pico a las 2 h y podría tardar horas en regresar a la situación basal.
Después de un periodo de varias horas de hipoxia grave persistente, la respuesta se vuelve aún más óptima y se duplica el incremento de la RVP antes de que ocurra una hipoxia prolongada[10 ].
Esta constricción exclusiva de la vasculatura pulmonar, es completamente diferente a la circulación sistémica, la cual responde con vasodilatación.
El mecanismo no está del todo claro. Se postulan varias teorías como la acidosis metabólica, la vasoconstricción por descarga simpática y la liberación de mediadores vasoconstrictores.
Los estudios más recientes indican la existencia de un incremento de la concentración del calcio iónico en el citoplasma por inhibición de los canales de potasio dependientes de voltaje y la despolarización de la membrana como consecuencia de tal. Esta teoría afirma que, ante una hipoxia, existe una estimulación de los sensores RedOx mitocondriales cuyos radicales libres actuarían como mediadores.
Además, en un estado hipóxico existe un incremento del gasto energético cuya alta demanda es compensada mediante glucolisis, donde la conversión enzimática de ADP a ATP resulta en un incremento de AMP. Esto activa las enzimas oxígeno-sensibles, terminando la reacción con liberación de Ca2+ desde el retículo sarcoplásmico.
En acidosis metabólica, la vasoconstricción es el resultado del aumento de la concentración de hidrogeniones a nivel extracelular. Por otra parte, modestos incrementos de la presión arterial de dióxido de carbono (PCO2 > 52 mmHg) producen aumento de la RVP[11 ], y aunque su respuesta sea lenta, logra un pico a las 4 h aproximadamente[12]. El efecto del dióxido de carbono (CO2) sobre la VPH está determinado por la presión alveolar de dióxido de carbono (PCO2) y la cantidad presente en la sangre venosa mixta. Se sugiere evitar hipocapnea en situaciones donde la VPH sea útil.
La hipotermia (incluso por debajo de 37 °C) en condiciones de normoxia, incrementa la VPH produciendo aumento del tono vascular pulmonar[13 ].
Un factor trascendental en la VPH es la cantidad de hierro plasmático. El hierro juega un papel muy importante en la atenuación de la VPH, pero también disminuye, en gran medida, la respuesta que se observa en la fase dos. La hipoferremia produce aumento de la presión en la arteria pulmonar (PAP) en pacientes con normoxia[14 ]. Sin embargo, la influencia exacta de la ferremia en la VPH en patología pulmonar, es desconocida. La hemooxigenasa es la enzima responsable de la degradación del grupo Hem y, además, es sensible al CO2, cuyo producto de degradación es el CO. El CO a su vez suprime la reactividad vascular pulmonar.
También se postula que ciertas sustancias vasoactivas que actúan a nivel del endotelio podrían jugar un papel trascendental. Un ejemplo de esto es el óxido nítrico inhalado (NOi)[15 ] usado como vasodilatador en concentraciones bajas (20 ppm).
La alteración del reflejo de VPH se ha estudiado en modelos experimentales, donde se plantean otras
teorías explicativas; como: la presencia de endotoxinas[16], la producción incrementada de prostanoides vasodilatadores[17], los productos de la ciclooxigenasa[18], la presencia de peróxido de hidrógeno[18] y últimamente se ha postulado la presencia del factor de activación plaquetario[19] como producto de la inflamación celular durante la injuria pulmonar aguda. Este daño incluye alteración endotelial, edema, vasoconstricción e incremento de la resistencia de la vía aérea pequeña[20 ].
En suma, como consecuencia de la hipoxemia, se produce una redistribución del flujo sanguíneo hacia las zonas mejor ventiladas, algo que podría denominarse “reclutamiento vascular” [21],[22 ].
Todo lo anteriormente expuesto son conceptos fisiológicos poco conocidos para quienes hacemos medicina intensiva del adulto. Sin embargo, los pediatras intensivistas lo han descrito muy bien y desde hace mucho tiempo atrás en relación a bronquiolitis y otras infecciones víricas[23 ].
En la vida fetal, la RVP es muy alta por dos razones: recibe únicamente 15% del volumen sistólico cardiaco y por obvia vasoconstricción hipóxica. Al nacer, con la primera ventilación, se oxigenan los alveolos lo cual produce una dramática disminución de la resistencia vascular pulmonar y, a consecuencia de la relajación del músculo liso vascular, hay un aumento de la producción de óxido nítrico (NO) y estimulación endotelial, lo que a su vez optimiza el flujo sanguíneo pulmonar. Lo mismo sucede en la enfermedad de membrana hialina, donde el uso de sustancias vasodilatadoras (NOi) produce sustancial mejoría del intercambio gaseoso por optimización de la relación ventilación-perfusión (V/Q)[24 ].
Similar situación ocurre con los recién nacidos que tienen hipertensión pulmonar basal y persistencia del foramen oval. En estos pacientes, los cambios en la presión de la arteria pulmonar inducidos por acidosis, producen incrementos del shunt intracardiaco y como resultado hay una alteración de la relación PaO2/FiO2[25]. Este deterioro podría magnificarse si se suma una alteración de la VPH a consecuencia de la inflamación local (por ejemplo: infecciones virales) y por factores inflamatorios propios de la respuesta inflamatoria sistémica.
La región anatómica exacta donde se produce el reflejo de VPH es incierta. La hipoxia disminuye el flujo vascular en las pequeñas arteriolas, arterias, capilares y vénulas pulmonares, y se cree que el mayor reflejo de VPH se produce a nivel de los capilares arteriales distales (< 200 um).
La visualización angiográfica ha demostrado que este efecto es mayor en los vasos proximales (bronquiolos terminales)[26 ], rompiendo el paradigma de exclusividad que se asumía en torno a los capilares alveolares.
Estudios experimentales aseguran, además, que existe algún grado de vasoconstricción venosa[27 ]. Sin embargo, la respuesta a la hipoxia también se produce en los capilares pulmonares que no tienen músculo liso como componente de su pared. En este caso, se plantea que la contracción probablemente se deba a la presencia de células de los septos interalveolares que producirían constricción capilar directa por distorsión de la anatomía alveolar[28 ]. Nuevamente, esto sería compatible con la hipótesis de Haldane[2 ].
La variabilidad en la intensidad de la respuesta está supeditada a varios factores; por ejemplo, ante reducciones graduales de PO2 se ha demostrado un incremento progresivo de la presión de arteria pulmonar. Mujeres usuarias de terapia de reemplazo hormonal también podrían experimentar una alteración en la magnitud de la vasoconstricción explicada por alteración hormonal de los componentes estructurales de la pared vascular.
El grado de redistribución del flujo vascular es inversamente proporcional al segmento hipóxico, es decir, mientras más pequeña sea la lesión hipóxica mayor será el porcentaje de redistribución de flujo vascular que se aleja de dicho segmento[29 ]. Esta redistribución del flujo vascular podría volverse menos efectiva ante grandes zonas hipóxicas[12 ] y la respuesta compensatoria consistiría en redistribuir el flujo hacia los ápices pulmonares a través del incremento de presión de la arteria pulmonar, venciendo el gradiente gravitacional. Este mecanismo adaptativo está presente tanto en pulmones sanos como en patológicos.
En este contexto, son interesantes los estudios que asocian al volutrauma con la disminución de la magnitud de respuesta del reflejo de VPH[30 ]. De hecho, se plantea que el aumento en la presión de la aurícula izquierda, probablemente empeorado por el uso de PEEP y aumento de la postcarga, contribuye al empeoramiento de la relación V/Q y la resultante hipoxemia[31 ].
La hipoxemia que se produce por atelectasias, es consecuencia de la alteración de la relación V/Q por persistencia de la perfusión en zonas no ventiladas. Factores mecánicos podrían contribuir a mantener la perfusión en estas zonas colapsadas. Sin embargo, la magnitud del reflejo de VPH variará según la producción de prostaglandinas vasodilatadoras en la zona atelectásica[32 ]. El factor inducible por hipoxia (HIF- 1), presente en casi todos los tejidos, se degrada (en normoxia) mediante hidroxilación. En hipoxia crece
de forma exponencial y activa factores citoprotectores que optimizan la oferta de oxígeno a los tejidos y mejoran su utilización.
El NO es una molécula regulada por el HIF-1[33] e inhibe el ciclo de replicación del coronavirus en el SDRA por SARS-CoV, lo cual fue demostrado en el 2005[34]. Aun no existen estudios concluyentes en relación SARS-CoV-2, pero podría ser que la acción inhibitoria del NO sobre el ciclo de replicación viral sea transversal para ambos tipos de coronavirus[34 ].
La fisiología de la hipoxia hipobárica (o hipoxia de la altitud) es muy similar a la del enfermo crítico[35 ]. A más de 8.000 metros de altura, con una presión parcial de oxígeno inspirado máxima de 43 mmHg, era muy difícil pensar que el ser humano lograría adaptarse a ese límite sin apoyo de oxígeno. Este mito se derrumbó en 1981 cuando se logró entender el concepto de la fisiología adaptativa[36 ]. La hipoxia, consecuencia de la disminución de la oferta de oxígeno, obliga a una adaptación metabólica constante para mantener la homeostasis energética[37 ]. Aclimatación es el término acuñado para explicar este fenómeno adaptativo que incluye cambios a nivel endocrinológico, cardíaco, hematológico, muscular, circulatorio y respiratorio (hiperventilación). El nexo de similitud entre la hipoxia hipobárica y el SDRA se estableció mediante análisis de muestras obtenidas por lavado broncoalveolar en las que encontraron factores proinflamatorios[33 ]. Además, se postula que existe un defecto del transporte de sodio transepitelial. Mediante el estudio por cateterismo de la arteria pulmonar se encontró que el aumento agudo de la presión capilar pulmonar (> 20 mmHg) ante la hipoxia, fue secundario a una vasoconstricción pulmonar[38 ] como consecuencia de la caída de la PO2 desde el aire inspirado hasta el interior de la mitocondria (cascada de oxígeno), afectando su biogénesis.
El reflejo de VPH también está presente en anfibios, peces, aves, reptiles y mamíferos[39 ]. Tiene un tiempo de respuesta inmediata (incluso a segundos después de presentarse la hipoxia) y esta respuesta puede prolongarse durante horas[40 ]. La hipoxia produce incrementos de presión de la arteria pulmonar sin que esto se traduzca en alteraciones de presión a nivel de la aurícula izquierda, ni tampoco que haya impacto sobre la frecuencia cardiaca. Sin embargo, se ha descrito repercusión variable sobre el volumen sistólico eyectado[41 ],[42].
-
Mediadores
La postulación se basa en la presencia de estímulos físicos y químicos que actuarían en los distintos re
ceptores de la pared vascular, destacando las catecolaminas, la histamina, la serotonina y la angiotensina II. Este último es un potente vasoconstrictor pulmonar en normoxia y podría atenuar la VPH mediante inactivación de la enzima convertidora de angiotensina II (ECA II)[43 ]. La ECA II es una enzima contrarreguladora que degrada la angiotensina II hasta angiotensina y ésta, a su vez, estimula la vasodilatación mediante la producción de óxido nítrico. También produce otros efectos como: atenuar el efecto vasoconstrictor de la angiotensina II, retener sodio y predisponer a la fibrosis[44 ]. Existe evidencia que demuestra que los pacientes con SARS-CoV-2 presentan niveles plasmáticos elevados de angiotensina II en relación directa con el daño pulmonar y la carga viral[45 ]. Esto se traduce en un aumento de la vasoconstricción pulmonar, del stress oxidativo, de la respuesta inflamatoria y procoagulante e incluso de la progresión hacia SDRA[46 ].
El sistema nervioso simpático tiene importante trascendencia en la mediación entre los quimiorreceptores y su nexo con el hipotálamo y el ganglio estrellado. Los antagonistas alfa adrenérgicos y agonistas beta adrenérgicos han sido descritos como probables mediadores atenuantes de la respuesta. Por el contrario, la vasoconstricción se ve potenciada por los antagonistas de los receptores beta[47 ]. La noradrenalina tiene un comportamiento bifásico: cuando el tono vascular es bajo actúa como vasoconstrictor y cuando el tono está aumentado produce vasodilatación[48 ].
Los agentes colinérgicos y no colinérgicos-adrenérgicos producen vasodilatación, sin llegar a tener un rol primario.
Una muy interesante explicación se basa en las vías metabólicas de la ciclooxigenasa y la lipooxigenasa como productos del metabolismo del ácido araquidónico[49 ,50], potenciando el reflejo de VPH ya que actuarían como moduladores y no como mediadores. Basados en este argumento, se ha experimentado con prostaglandinas y leucotrienos como inhibidores de la VPH.
Evidencia reciente ha demostrado la existencia de péptidos vasoactivos a nivel endotelial, destacando la endotelina como factor vasoconstrictor y el NO como el mayor representante del efecto dilatador. La producción endotelial basal de NO permite mantener la circulación pulmonar en un estado activo de vasodilatación. La óxido nítrico sintasa (NOs) es la enzima responsable de la síntesis de NO a partir de L-arginina. En condiciones de hipoxia, esta enzima se inactiva.
-
Modelo de respuesta
– El estímulo[51 ]. En el pulmón ventilado, la presión alveolar de oxígeno (PAO2) será siempre mayor a la PvO2, por tanto:
PestímuloO2 = PAO2 0,62 + PvO2 0,38
• Esta ecuación se obtuvo de manera experimental en animales. Fisiológicamente no habría argumento para refutar su aplicabilidad en humanos[51 ].
• Los cambios de PvO2 presentes en situaciones como atelectasias, pacientes monopulmonares, cambios en el volumen sistólico, consumo de oxígeno y vasodilatación o vasoconstricción, afectarán directamente el reflejo de VPH.
– El sensor de la hipoxia es la arteriola precapilar. Incluye el sistema de fosforilación oxidativa a nivel mitocondrial y la vía de la citocromo P450[52].
– El transductor. Representado por la membrana electrofisiológica.
– El efector son las células de la musculatura lisa vascular. Aunque el mecanismo mediante el cual se produce el efecto de VPH aún no está del todo claro, se plantea la teoría de que la hipoxia incrementaría la permeabilidad al calcio iónico desde el espacio extracelular hacia el espacio intracelular. El resultante aumento de su concentración explicaría la despolarización eléctrica y la constricción[53 ]. Paralelo a esto, también se plantea la acción que tendrían los radicales libres de oxígeno, directa e indirectamente sobre los canales de calcio[54 ].
-
Relación V/Q
En el parénquima pulmonar, la PO2 local está determinada por el cociente entre la oferta de oxígeno al tejido pulmonar (ventilación alveolar o VA) y la entrega de oxígeno desde los pulmones a los tejidos sistémicos o perfusión (Q):
PO2 = VA/Q
En los tejidos sistémicos, la PO2 local está determinada por el ratio entre la oferta de oxígeno a los tejidos o perfusión (Q) y el consumo local de oxígeno (VO2):
PO2 = Q/VO2
El equilibrio ventilación/perfusión (V/Q) es el factor fundamental para optimizar el intercambio gaseoso (Figura 1 ). En condiciones de normoxia el ratio V/Q es igual a 1, con aproximadamente 5 L/min de flujo vascular. El reflejo de VPH disminuye el flujo vascular en las áreas pulmonares donde la PAO2 es baja (V/Q bajo), redistribuyendo este flujo hacia zonas con mejor ventilación (V). Con PO2 menor a 25 mmHg (alveolar y venosa mixta) la VPH se vuelve menos efectiva[55 ] y esto es evidente en pacientes con patología pulmonar.
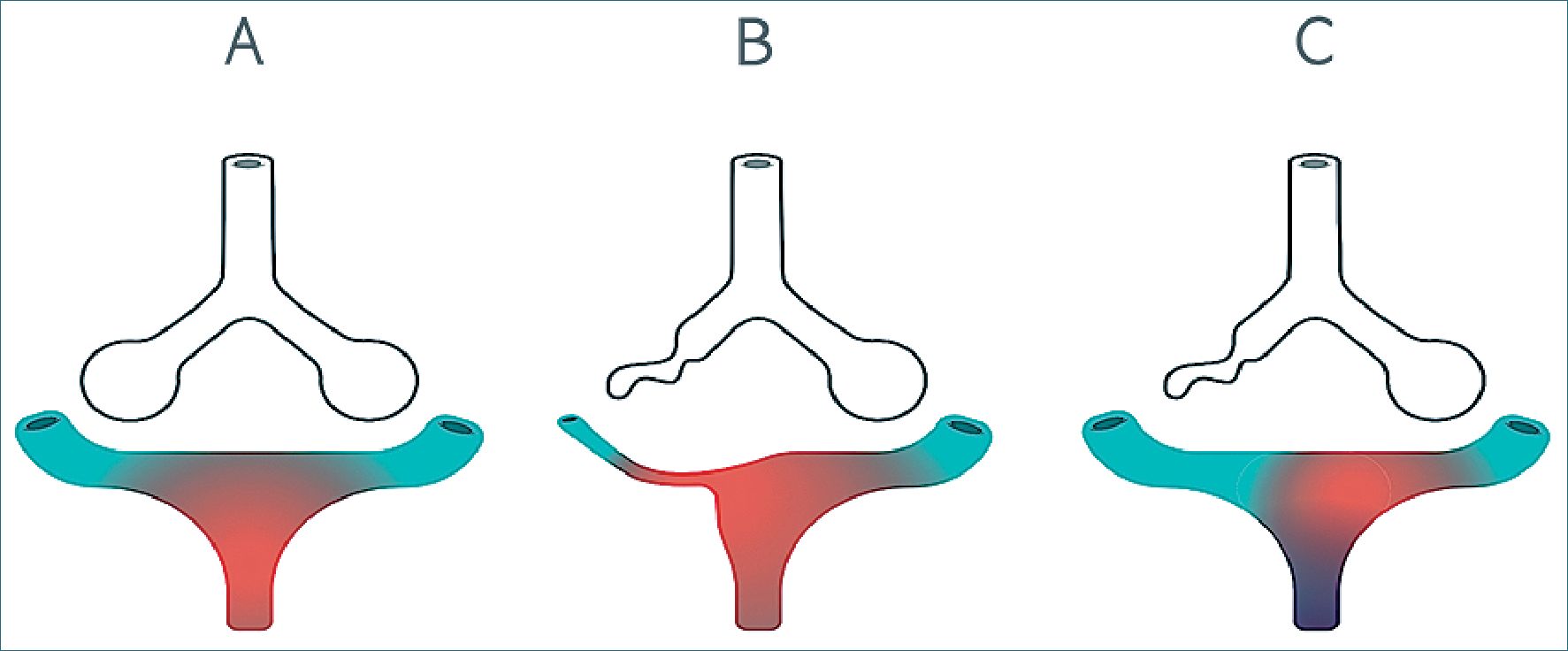
Figura 1. Mecanismo del reflejo de VPH. WeVent 2020.
A) Situación Normal. El flujo vascular en todas las regiones pulmonares es completamente oxigenado en el alveolo. V/Q = 1. B) Reflejo fisiológico de VPH. Ante una unidad alveolar pobremente ventilada, la VPH redirecciona el flujo vascular. Esto aumenta el ratio V/Q y, como consecuencia, aumenta la PaO2. V/Q < 1.
C) Pérdida del reflejo de VPH. Ante un área no ventilada, la PO2 de esta región es igual a la de la sangre venosa mixta. Ante una hipoxia tisular se pierde el reflejo de VPH provocando vasodilatación y magnificando el shunt. V/Q = 0.
El tamaño de la región con ratio V/Q baja juega un papel trascendental en la capacidad del reflejo de VPH para mejorar la oxigenación, siendo el valor máximo de tolerancia entre 30% y 70% de pulmón hipóxico. A partir de este rango, el reflejo se vuelve menos efectivo.
En un pulmón patológico que presenta una hipoxia por colapso pulmonar, la VPH podría reducir el flujo vascular hasta en 50% en la zona afectada[56 ]. Ante una injuria aguda la mayor barrera fisiopatológica para la oxigenación es el shunt derecha a izquierda y la VPH contribuye a su reducción. Cuando se administra una elevada fracción inspirada de oxígeno (FiO2) en pulmones pobremente ventilados, la disminución de la VPH produce un incremento de la mezcla venosa y un aumento del espacio muerto en las unidades alveolares bien ventiladas[57 ]. Este aumento del shunt podría no verse reflejado, desde el punto de vista clínico, cuando se aumenta la FiO2 por obvia mejoría de la saturación periférica. Entonces, en etapas tempranas, cuando la hipoxia es compensada por el reflejo de VPH, el paciente no presenta apremio respiratorio. Bajo estas circunstancias no parece muy acertada la denominación de “hipoxemia feliz”[58 ]. La complianza (Crs) preservada está determinada por la existencia de áreas pulmonares bien ventiladas y, además, explica la eupnea del paciente en estadio temprano de la enfermedad[59 ].
Sin embargo, el incremento del espacio muerto puede aumentar la PaCO2 si el paciente o el especialista sanitario encargado del soporte ventilatorio no logran aumentar la ventilación alveolar. Esto se compensa con taquipnea o aumento de la frecuencia respiratoria y con hiperpnea o aumento del volumen corriente (Vc).
El objetivo, sin duda, será disminuir la PaCO2 mediante la hiperventilación, aunque esto producirá disminución de la vasoconstricción y empeorará la situación.
Este incremento forzado y compensatorio del Vc produce un aumento secuencial de la presión negativa intratorácica. Si a esto se suma la historia natural de progresión de la enfermedad (inflamación, aumento de la permeabilidad vascular por vasoplejia e hiperadrenergia) seguramente se generará un acelerador de la lesión pulmonar (strain raiser) en el contexto de la lesión pulmonar autoinflingida (P-SILI)[60 ,61]. La persistencia de esto derivará en aumento del shunt e hipoxemia (fase tardía o SDRA clásico). La actividad protrombótica y el estado procoagulante, son un importante contribuyente para el disbalance del ratio V/Q[62,63]. Se recomienda la medición del gradiente alveolo-arterial (A-a), el cual se incrementa por la alteración del ratio V/Q.
-
Enfoque terapéutico
La respuesta vasoconstrictora en el SDRA representa una alteración elemental de la vasorreactividad pulmonar (Figura 2 ).
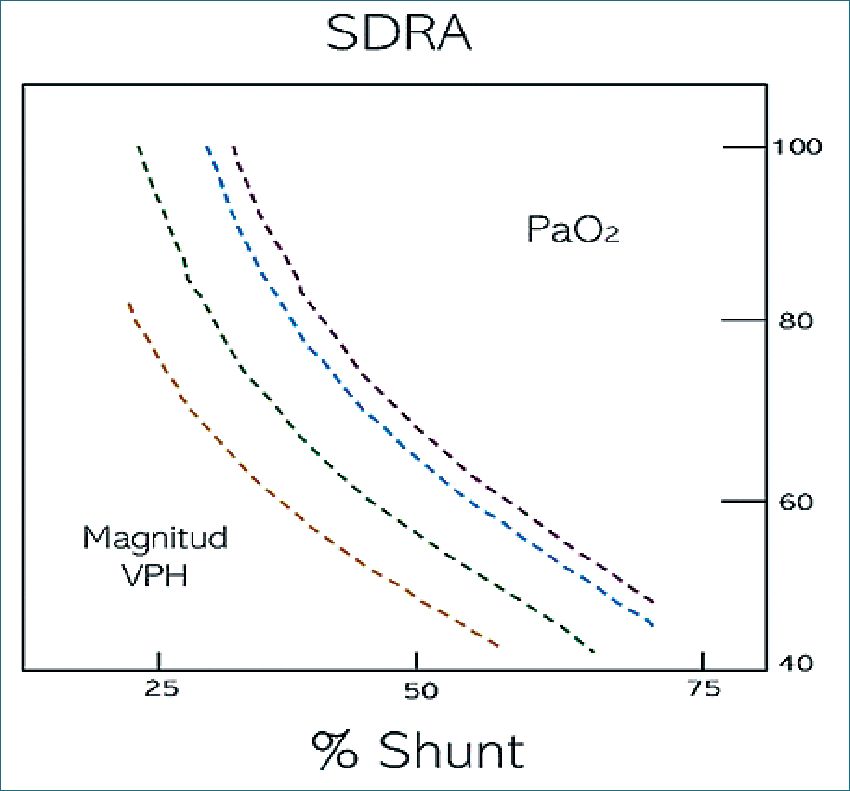
Figura 2. En pacientes con SDRA, la hipoxemia resultante del aumento del shunt intrapulmonar, podría ser compensada transitoriamente por el reflejo de VPH. Adaptado (con permiso) de Brimioulle S, Lejeune P, Naeije R: Effects of hypoxic pulmonary vasoconstriction on gas exchange. J Appl Physiol 1996, 81: 1535-1543.
Los pacientes con SDRA tienen hipoxemia como consecuencia de un aumento del shunt[64 ],[65], pero una vigorosa VPH podría incrementar hasta 20 mmHg la PaO2[65].
Se han desarrollado diversos estudios para optimizar el “reclutamiento vascular”. En adultos con SDRA se logró optimizar la distribución del flujo vascular mediante la aplicación simultánea de sustancias vasodilatadoras y vasoconstrictoras[66 ]. La almitrina produce vasoconstricción de la totalidad del territorio pulmonar, incluyendo los territorios mal ventilados. Si a esto se suma el efecto vasodilatador del NOi[67 ] en las zonas ventiladas, se logra amplificar y optimizar el “reclutamiento vascular”, optimizando la perfusión hacia las zonas mejor ventiladas. El efecto equivalente de la almitrina se logra con la noradrenalina, siendo este último el que tiene mejor perfil de seguridad y aplicabilidad. Si a estos dos vasoactivos, se suma el impacto del prono, el resultado es mucho más importante[68 ].
Sabemos que, en un paciente que presenta hipoxemia por SDRA y que requiere un soporte ventilatorio invasivo, la PEEP juega un rol trascendental reduciendo el shunt. Con una programación adecuada y personalizada de la PEEP se evitará el colapso de las unidades alveolares y esto aumentará la capacidad residual funcional (CRF), que a su vez mejorará el equilibrio V/Q[69]. El uso de la PEEP podría optimizar la respuesta de VPH al mejorar la oxigenación y contribuir a la redistribución del flujo vascular por efecto mecánico.
La alteración de la perfusión pulmonar en relación a SARS-CoV-2, ha sido corroborada mediante técnicas especializadas de imágenes[70 ],[71]. La convivencia de zonas con ratio V/Q normal o alto (parénquima sano con flujo aumentado por la vasoconstricción) y zonas con ratio V/Q bajo (parénquima enfermo con flujo vascular disminuido por la vasoplejia), representaría la etapa de transición entre estadios[72 ]. Esto explica el motivo por el cual en posición prono hay mejoría de la hipoxemia.
-
Mensaje final
Con base en lo expuesto, ante un paciente que presenta una hipoxia, deberíamos analizar el shunt mediante el uso de la relación PaO2/FiO2. Una forma práctica de valorar esto, es mediante una radiografía de tórax. Esto será de especial importancia en un paciente con SDRA y en particular, tal como se ha constatado durante esta pandemia, en los pacientes con infecciones víricas como la causada por el SARS- CoV-2.
Ante una hipoxemia grave (PaO2/FiO2 < 200 que representa un shunt superior al 30%) y con una radiografía de tórax (o con TAC) sin claros y extensos infiltrados bilaterales, el shunt podría deberse a una afectación vascular. Esto podría explicarse por dos causas:
1. Un aumento de la RVP, que podría generar un shunt derecha a izquierda a través del formane oval permeable, cuya prevalencia es de hasta el 20% en pacientes adultos con SDRA[73 ].
2. Una alteración del reflejo de VPH.
En ambos casos, tratar de redistribuir el flujo vascular con el uso concomitante de vasoconstrictores pulmonares y de NOi podría ser de utilidad, aunque hace falta una evaluación con ensayos clínicos. Además, en esta fase de la enfermedad, el uso de PEEP alta (10 a 15 cmH2O) podría ser contraproducente ya que podría aumentar la sobredistensión (ya que la Crs puede ser normal) y también podría aumentar el shunt y el espacio muerto.
Es importante destacar que si el cuadro evoluciona hacia un SDRA clásico, el NOi no tendrá ninguna utilidad y podría aumentar la mortalidad por aumento de la incidencia de fracaso renal. Para enfrentar la hipoxia, no se trata de aumentar el suministro de oxígeno sino de mejorar su utilización[74 ]-[76]. Históricamente hemos aceptado la hipercapnea permisiva y ahora debemos aceptar la hipoxia adaptativa ante la injuria pulmonar, la cual será inicialmente compensada por la activación del reflejo de VPH.
Referencias
1. Euler US, Liljestrand G; Von Euler et al. Observation on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12(4):301–20. https://doi.org/10.1111/j.1748-1716.1946.tb00389.x.
2. – Haldane JS. Respiration. New Haven: Yale University Press; 1922. p. 427.
3. – Gattinoni L, Coppola S, Chiumello D. ¨Repply to: Hedenstierna et al, Haouzi et al, Maley et al, Fowler et al, Bathia and Mohammed, Bos & Koumbourlis and Motoyama. ¨American Journal of Respiratory and Critical Care, 0(ja), pp. June 24, 2020. https://doi.org/10.1164/rccm.202004-1052LE.
4. Gattinoni L, Chiumello D, Caironi P, Busana M, Romitti F, Brazzi L, et al. COVID-19 pneumonia: different respiratory treatments for different phenotypes? Intensive Care Med. 2020 Jun;46(6):1099–102. https://doi.org/10.1007/s00134-020-06033-2 PMID:32291463
5. Camporota L, Vasques F, Sanderson B, Barrett NA, Gattinoni L. Identification of pathophysiological patterns for triage and respiratory support in COVID-19. Lancet Respir Med. 2020 Aug;8(8):752–4. https://doi.org/10.1016/S2213-2600(20)30279-4 PMID:32598907
6. Fan E, Beitler JR, Brochard L, Calfee CS, Ferguson ND, Slutsky AS, et al. COVID-19-associated acute respiratory distress syndrome: is a different approach to management warranted? Lancet Respir Med. 2020 Aug;8(8):816–21. https://doi.org/10.1016/S2213-2600(20)30304-0 PMID:32645311
7. Hedenstierna G, Chen L, Hedenstierna M, Lieberman R, Fine DH. Nitric oxide dosed in short bursts at high concentrations may protect against Covid 19. Nitric Oxide. 2020 Oct;103:1–3. https://doi.org/10.1016/j.niox.2020.06.005 PMID:32590117
8. West JB. Fisiología respiratoria.8.ª ed. Editorial Lippincott Williams and Walters Kluver, 2009.53-54.
9. Talbot NP, Balanos GM, Dorrington KL, Robbins PA. Two temporal components within the human pulmonary vascular response to approximately 2 h of isocapnic hypoxia. J Appl Physiol (1985). 2005 Mar;98(3):1125–39. https://doi.org/10.1152/japplphysiol.00903.2004 PMID:15542574
10. – Smith TG, Balanos GM, Croft QP, Talbot NP, Dorrington KL, Ratcliffe PJ, Robbins PA. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J Physiol 2008; 586(Pt 24):5999–6005 11.- Kiely DG, Cargill RI, Lipworth BJ: Effects of hypercapnia on hemodynamic, inotropic, lusitropic, and electrophysiologic indices in humans. Chest. 1996;109:1215–21.
12. – Balanos GM, Talbot NP, Dorrington KL, Robbins PA. Human pulmonary vascular response to 4h of hypercapnia and hypocapnia measured using Doppler echocardiography. J Appl Physiol. 1985;2003(94):1543–51.
13. Benumof JL, Wahrenbrock EA. Dependency of hypoxic pulmonary vasoconstriction on temperature. J Appl Physiol. 1977 Jan;42(1):56–8. https://doi.org/10.1152/jappl.1977.42.1.56 PMID:833077
14. – Balanos GM, Dorrington KL, Robbins PA: Desferrioxamine elevates pulmonary vascular resistance in humans: Potential for involvement of HIF-1. J Appl Physiol (1985) 2002; 92:2501–7, pp 341-50
15. Benzing A, Mols G, Brieschal T, Geiger K. Hypoxic pulmonary vasoconstriction in nonventilated lung areas contributes to differences in hemodynamic and gas exchange responses to inhalation of nitric oxide. Anesthesiology. 1997 Jun;86(6):1254–61. https://doi.org/10.1097/00000542-199706000-00005 PMID:9197293
16. Benumof JL, Wahrenbrock EA. Blunted hypoxic pulmonary vasoconstriction by increased lung vascular pressures. J Appl Physiol. 1975 May;38(5):846–50. https://doi.org/10.1152/jappl.1975.38.5.846 PMID:1126894
17. Garrett RC, Thomas HM 3rd. Meclofenamate uniformly decreases shunt fraction in dogs with lobar atelectasis. J Appl Physiol. 1983 Jan;54(1):284–9. https://doi.org/10.1152/jappl.1983.54.1.284 PMID:6826414
18. Sylvester JT, Shimoda LA, Aaronson PI, Ward JP. Hypoxic pulmonary vasoconstriction. Physiol Rev. 2012 Jan;92(1):367–520. https://doi.org/10.1152/physrev.00041.2010 PMID:22298659
19. Domino KB, Wetstein L, Glasser SA, Lindgren L, Marshall C, Harken A, et al. Influence of mixed venous oxygen tension (PVO2) on blood flow to atelectatic lung. Anesthesiology. 1983 Nov;59(5):428–34. https://doi.org/10.1097/00000542-198311000-00012 PMID:6638551
20. Simpson SQ. Oxygen-induced acute hypercapnia in chronic obstructive pulmonary disease: what’s the problem? Crit Care Med. 2002 Jan;30(1):258–9. https://doi.org/10.1097/00003246-200201000-00045 PMID:11902279
21. Wagner WW Jr, Latham LP, Capen RL. Capillary recruitment during airway hypoxia: role of pulmonary artery pressure. J Appl Physiol. 1979 Aug;47(2):383–7. https://doi.org/10.1152/jappl.1979.47.2.383 PMID:468695
22. Capen RL, Wagner WW Jr. Intrapulmonary blood flow redistribution during hypoxia increases gas exchange surface area. J Appl Physiol. 1982 Jun;52(6):1575–80. https://doi.org/10.1152/jappl.1982.52.6.1575 PMID:7201993
23. Almeida-Junior AA, da Silva MT, Almeida CC, Ribeiro JD. Relationship between physiologic deadspace/tidal volume ratio and gas exchange in infants with acute bronchiolitis on invasive mechanical ventilation. Pediatr Crit Care Med. 2007 Jul;8(4):372–7. https://doi.org/10.1097/01.PCC.0000269389.51189.A8 PMID:17545938
24. Kinsella JP, Ivy DD, Abman SH. Inhaled nitric oxide improves gas exchange and lowers pulmonary vascular resistance in severe experimental hyaline membrane disease. Pediatr Res. 1994 Sep;36(3):402–8. https://doi.org/10.1203/00006450-199409000-00022 PMID:7808839
25. – Rudolph AM, Yuan S, Einstein A, Rd E, Ave MP, Rudolph AM, et al. Response of the Pulmonary Vasculature to Hypoxia and H + Ion Concentration Changes * of hypoxia on the pulmonary vasculature has. 1966;45(3)
26. Kato M, Staub NC. Response of small pulmonary arteries to unilobar hypoxia and hypercapnia. Circ Res. 1966 Aug;19(2):426–40. https://doi.org/10.1161/01.RES.19.2.426 PMID:5914855
27. Morgan BC, Church SC, Guntheroth WG. Hypoxic constriction of pulmonary artery and vein in intact dogs. J Appl Physiol. 1968;25(4):356–61. https://doi.org/10.1152/jappl.1968.25.4.356.
28. Kapanci Y, Assimacopoulos A, Irle C, Zwahlen A, Gabbiani G. “Contractile interstitial cells” in pulmonary alveolar septa: a possible regulator of ventilation-perfusion ratio? Ultrastructural, immunofluorescence, and in vitro studies. J Cell Biol. 1974 Feb;60(2):375–92. https://doi.org/10.1083/jcb.60.2.375 PMID:4204972
29. Scanlon TS 3rd, Benumof JL, Wahrenbrock EA, Nelson WL. Hypoxic pulmonary vasoconstriction and the ratio of hypoxic lung to perfused normoxic lung. Anesthesiology. 1978 Sep;49(3):177–81. https://doi.org/10.1097/00000542-197809000-00005 PMID:686439
30. – Zapol WM, Rie MA, Frikker M. Pulmonary circulation during adult respiratory distress syndrome. In: Zapol WM, Falke KJ, editors. Acute respiratory failure. New York; 1985. pp. 241–71.
31. Reeves JT, Grover RF. Blockade of acute hypoxic pulmonary hypertension by endotoxin. J Appl Physiol. 1974 Mar;36(3):328–32. https://doi.org/10.1152/jappl.1974.36.3.328 PMID:4592254
32. Hales CA, Sonne L, Peterson M, Kong D, Miller M, Watkins WD. Role of thromboxane and prostacyclin in pulmonary vasomotor changes after endotoxin in dogs. J Clin Invest. 1981 Aug;68(2):497–505. https://doi.org/10.1172/JCI110281 PMID:7021593
33. Kubo K, Hanaoka M, Yamaguchi S, Hayano T, Hayasaka M, Koizumi T, et al. Cytokines in bronchoalveolar lavage fluid in patients with high altitude pulmonary oedema at moderate altitude in Japan. Thorax. 1996 Jul;51(7):739–42. https://doi.org/10.1136/thx.51.7.739 PMID:8882083
34. Avellanas Chavala ML. Un viaje entre la hipoxia de la gran altitud y la hipoxia del enfermo crítico : ¿ qué puede enseñarnos en la compresión y manejo de las enfermedades críticas? Med Int. 2018;42(6):380–90. https://doi.org/10.1016/j.medin.2017.08.006.
35. West JB. Career perspective: John B West. Extrem Physiol Med. 2012 Nov;1(1):11–4. https://doi.org/10.1186/2046-7648-1-11 PMID:23849052
36. Raguso CA, Luthy C. Nutritional status in chronic obstructive pulmonary disease: role of hypoxia. Nutrition. 2011 Feb;27(2):138–43. https://doi.org/10.1016/j.nut.2010.07.009 PMID:21145207
37. Akerström S, Mousavi-Jazi M, Klingström J, Leijon M, Lundkvist A, Mirazimi A. Nitric oxide inhibits the replication cycle of severe acute respiratory syndrome coronavirus. J Virol. 2005 Feb;79(3):1966–9. https://doi.org/10.1128/JVI.79.3.1966-1969.2005 PMID:15650225
38. Maggiorini M, Mélot C, Pierre S, Pfeiffer F, Greve I, Sartori C, et al. High-altitude pulmonary edema is initially caused by an increase in capillary pressure. Circulation. 2001 Apr;103(16):2078–83. https://doi.org/10.1161/01.CIR.103.16.2078 PMID:11319198
39. – Fishman A. Pulmonary circulation. IN: Handbook of physiology, Fishman AP, ed. Section 3: The respiratory system, Vol I: Circulation and nonrespiratory functions, chap 3, Baltimore: Williams and Wilkins, 1985:93-167
40. Malik AB, Kidd BS. Time course of pulmonary vascular response to hypoxia in dogs. Am J Physiol. 1973 Jan;224(1):1–6. https://doi.org/10.1152/ajplegacy.1973.224.1.1 PMID:4683301
41. Koyama T, Nakajima S, Kakiuchi Y. Quick increase of pulmonary blood flow in response to an acute alveolar hypoxia in human subjects. Jpn J Physiol. 1977;27(1):1–11. https://doi.org/10.2170/jjphysiol.27.1 PMID:864865
42. Motley HL, Cournand A, Werko L, Himmelstein A, Dresdale D. The influence of short periods of induced acute anoxia upon pulmonary artery pressures in man. Am J Physiol. 1947 Aug;150(2):315–20. https://doi.org/10.1152/ajplegacy.1947.150.2.315 PMID:20258388
43. Cargill RI, Lipworth BJ. Acute effects of hypoxaemia and angiotensin II in the human pulmonary vascular bed. Pulm Pharmacol. 1994 Oct;7(5):305–10. https://doi.org/10.1006/pulp.1994.1036 PMID:7626917
44. Vaduganathan M, Vardeny O, Michel T, McMurray JJ, Pfeffer MA, Solomon SD. Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med. 2020 Apr;382(17):1653–9. https://doi.org/10.1056/NEJMsr2005760 PMID:32227760
45. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020 Mar;63(3):364–74. https://doi.org/10.1007/s11427-020-1643-8 PMID:32048163
46. Zhang H, Baker A. Recombinant human ACE2: acing out angiotensin II in ARDS therapy. Crit Care. 2017 Dec;21(1):305. https://doi.org/10.1186/s13054-017-1882-z PMID:29237475
47. Porcelli RJ, Viau AT, Naftchi NE, Bergofsky EH. beta-Receptor influence on lung vasoconstrictor responses to hypoxia and humoral agents. J Appl Physiol. 1977 Oct;43(4):612–6. https://doi.org/10.1152/jappl.1977.43.4.612 PMID:198395
48. Silove ED, Inoue T, Grover RF. Comparison of hypoxia, pH, and sympathomimetic drugs on bovine pulmonary vasculature. J Appl Physiol. 1968 Mar;24(3):355–65. https://doi.org/10.1152/jappl.1968.24.3.355 PMID:5640722
49. Weir EK, McMurtry IF, Tucker A, Reeves JT, Grover RF. Prostaglandin synthetase inhibitors do not decrease hypoxic pulmonary vasoconstriction. J Appl Physiol. 1976 Nov;41(5 Pt. 1):714–8. https://doi.org/10.1152/jappl.1976.41.5.714 PMID:825505
50. Voelkel NF, Gerber JG, McMurtry IF, Nies AS, Reeves JT. Release of vasodilator prostaglandin, PGI2, from isolated rat lung during vasoconstriction. Circ Res. 1981 Feb;48(2):207–13. https://doi.org/10.1161/01.RES.48.2.207 PMID:7006847
51. – Marshall BE, Marshall C, Frasch HF (Stanley TH, Sperry RJ, editors). Control of the pulmonary circulation, Anesthesia and the Lung. Dordrecht: Kluwer; 1992. pp. 9–18.
52. Miller MA, Hales CA. Role of cytochrome P-450 in alveolar hypoxic pulmonary vasoconstriction in dogs. J Clin Invest. 1979 Aug;64(2):666–73. https://doi.org/10.1172/JCI109507 PMID:457876
53. McMurtry IF. BAY K 8644 potentiates and A23187 inhibits hypoxic vasoconstriction in rat lungs. Am J Physiol. 1985 Oct;249(4 Pt 2):H741–6. PMID:2413777
54. Archer SL, Will JA, Weir EK. Redox status in the control of pulmonary vascular tone. Herz. 1986 Jun;11(3):127–41. PMID:3017827
55. Burghuber O, Mathias MM, McMurtry IF, Reeves JT, Voelkel NF. Lung edema due to hydrogen peroxide is independent of cyclooxygenase products. J Appl Physiol. 1984 Apr;56(4):900–5. https://doi.org/10.1152/jappl.1984.56.4.900 PMID:6427146
56. Gillespie MN, Bowdy BD. Impact of platelet activating factor on vascular responsiveness in isolated rat lungs. J Pharmacol Exp Ther. 1986 Feb;236(2):396–402. PMID:3944767
57. Hamasaki Y, Mojarad M, Saga T, Tai HH, Said SI. Platelet-activating factor raises airway and vascular pressures and induces edema in lungs perfused with platelet-free solution. Am Rev Respir Dis. 1984 May;129(5):742–6. https://doi.org/10.1164/arrd.1984.129.5.742 PMID:6547031
58. Dhont S, Derom E, Van Braeckel E, Depuydt P, Lambrecht BN. The pathophysiology of ‘happy’ hypoxemia in COVID-19. Respir Res. 2020 Jul;21(1):198. https://doi.org/10.1186/s12931-020-01462-5 PMID:32723327
59. – Ottestad W, Seim M, Mæhlen JO. Covid-19 med stille hypoksemi. Tidsskr Nor LaegeforenNLM (Medline). 2020;140. Epub ahead of print.
60. – Barach AL, Martin J, Eckman M. Positive pressure respiration and its application to the treatment of acute pulmonary edema. Ann Intern
61. – Komorowski M, Aberegg SK. Using applied lung physiology to understand COVID-19 patterns. Br J Anaesth. 2020;0 Elsevier; [cited 2020 May 31]. https://linkinghub.elsevier.com/retrieve/pii/S0007091220303755
62. – Monsalve-Naharro JÁ, Domingo-Chiva E, Castillo SG, Cuesta-Montero P, Jiménez-Vizuete JM. Inhaled nitric oxide in adult patients with acute respiratory distress syndrome. Farm HospSociedad Espanola de Farmacia Hospitalaria. 2017:292–312. Epub ahead of print.
63. – Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. 2020;jth.14872 [cited 2020 Apr 25]. https://onlinelibrary.wiley.com/doi/abs/10.1111/jth.14872
64. Quebbeman EJ, Dawson CA. Effect of lung inflation and hypoxia on pulmonary arterial blood volume. J Appl Physiol. 1977 Jul;43(1):8–13. https://doi.org/10.1152/jappl.1977.43.1.8 PMID:583630
65. Mélot C, Lejeune P, Leeman M, Moraine JJ, Naeije R. Prostaglandin E1 in the adult respiratory distress syndrome. Benefit for pulmonary hypertension and cost for pulmonary gas exchange. Am Rev Respir Dis. 1989 Jan;139(1):106–10. https://doi.org/10.1164/ajrccm/139.1.106 PMID:2912329
66. – Bazin JE, Mansoor O. Thierry GiUart MD, Jean E. Bazin. 1998;402–9.
67. Afshari A, Brok J, Møller AM, Wetterslev J. Inhaled nitric oxide for acute respiratory distress syndrome and acute lung injury in adults and children: a systematic review with meta-analysis and trial sequential analysis. Anesth Analg. 2011 Jun;112(6):1411–21. https://doi.org/10.1213/ANE.0b013e31820bd185 PMID:21372277
68. Papazian L, Bregeon F, Gaillat F, Thirion X, Roch A, Cortes E, et al. Inhaled NO and almitrine bismesylate in patients with acute respiratory distress syndrome: effect of noradrenalin. Eur Respir J. 1999 Dec;14(6):1283–9. https://doi.org/10.1183/09031936.99.14612839 PMID:10624756
69. – Unzueta MC. Ventilación mecánica en cirugía torácica. In: Belda FJ, Lloréns J, editors. Ventilación mecánica en anestesia y cuidados críticos. España: Arán Ediciones S.L.; 2009. pp. 573–93.
70. – Lang M, Som A, Mendoza DP, Flores EJ, Reid N, Carey D, et al. Hypoxaemia related to COVID-19: vascular and perfusion abnormalities on dual-energy CT. Lancet Infect Dis. 2020;S1473-3099(20):30367–4. . https://doi.org/10.1016/S1473-3099(20)30367-4.
71. Grob D, Smit E, Prince J, Kist J, Stöger L, Geurts B, et al. Iodine maps from subtraction CT or dual-energy CT to detect pulmonary emboli with CT angiography: a multiple-observer study. Radiology. 2019 Jul;292(1):197–205. https://doi.org/10.1148/radiol.2019182666 PMID:31084482
72. Santamarina MG, Boisier D, Contreras R, Baque M, Volpacchio M, Beddings I. COVID-19: a hypothesis regarding the ventilation-perfusion mismatch. Crit Care. 2020 Jul;24(1):395. https://doi.org/10.1186/s13054-020-03125-9 PMID:32631389
73. Mekontso Dessap A, Boissier F, Leon R, Carreira S, Campo FR, Lemaire F, et al. Prevalence and prognosis of shunting across patent foramen ovale during acute respiratory distress syndrome. Crit Care Med. 2010 Sep;38(9):1786–92. https://doi.org/10.1097/CCM.0b013e3181eaa9c8 PMID:20601861
74. Helmerhorst HJ, Schultz MJ, van der Voort PH, de Jonge E, van Westerloo DJ. Bench-to-bedside review: the effects of hyperoxia during critical illness. Crit Care. 2015 Aug;19(1):284. https://doi.org/10.1186/s13054-015-0996-4 PMID:26278383
75. Six S, Jaffal K, Ledoux G, Jaillette E, Wallet F, Nseir S. Hyperoxemia as a risk factor for ventilator-associated pneumonia. Crit Care. 2016 Jun;20(1):195. https://doi.org/10.1186/s13054-016-1368-4 PMID:27334713
76. Vincent JL, Taccone FS, He X. Harmful effects of hyperoxia in postcardiac arrest, sepsis traumatic brain injury, or stroke: the importance of individualized oxygen therapy in critically ill patients. Can Respir J. 2017;2017:2834956. https://doi.org/10.1155/2017/2834956 PMID:28246487